В химии гипервалентная молекула (это явление иногда в разговорной речи называют расширенным октетом ) — это молекула , которая содержит один или несколько элементов главной группы, по -видимому, имеющих более восьми электронов в своих валентных оболочках . Пентахлорид фосфора ( PCl5 ), гексафторид серы ( SF6 ) , трифторид хлора ( ClF3 ), хлорит ( ClO−2) ион в хлористой кислоте и трииодид ( I−3) ионы являются примерами гипервалентных молекул.
Гипервалентные молекулы были впервые формально определены Джереми И. Мушером в 1969 году как молекулы, имеющие центральные атомы групп 15–18 в любой валентности , кроме самой низкой (т. е. 3, 2, 1, 0 для групп 15, 16, 17, 18 соответственно, на основе правила октета ). [1]
Существует несколько конкретных классов гипервалентных молекул:
Номенклатура NXL, введенная совместно исследовательскими группами Мартина , Ардуенго и Кочи в 1980 году [2] , часто используется для классификации гипервалентных соединений элементов главной группы, где:
Примеры номенклатуры NXL включают в себя:
Дебаты о природе и классификации гипервалентных молекул восходят к Гилберту Н. Льюису и Ирвингу Ленгмюру и дебатам о природе химической связи в 1920-х годах. [3] Льюис поддерживал важность двухцентровой двухэлектронной (2c-2e) связи в описании гипервалентности, таким образом, используя расширенные октеты для учета таких молекул. Используя язык орбитальной гибридизации, связи молекул, таких как PF 5 и SF 6 , как было сказано, были построены из sp 3 d n орбиталей на центральном атоме. Ленгмюр, с другой стороны, поддерживал доминирование правила октета и предпочитал использование ионных связей для учета гипервалентности, не нарушая правила (например, " SF2+
42F − " для SF 6 ).
В конце 1920-х и 1930-х годов Сагден утверждал о существовании двухцентровой одноэлектронной (2c-1e) связи и таким образом рационализировал связь в гипервалентных молекулах без необходимости расширенных октетов или характера ионной связи; в то время это было плохо принято. [3] В 1940-х и 1950-х годах Рандл и Пиментел популяризировали идею трехцентровой четырехэлектронной связи , которая по сути является той же концепцией, которую Сагден пытался развить десятилетиями ранее; трехцентровую четырехэлектронную связь можно альтернативно рассматривать как состоящую из двух коллинеарных двухцентровых одноэлектронных связей, при этом оставшиеся два несвязывающих электрона локализованы в лигандах. [3]
Попытка фактически приготовить гипервалентные органические молекулы началась с Германа Штаудингера и Георга Виттига в первой половине двадцатого века, которые стремились бросить вызов существующей теории валентности и успешно приготовить гипервалентные молекулы с центром в азоте и фосфоре. [4] Теоретическая основа гипервалентности не была очерчена до работы Дж. И. Мушера в 1969 году. [1]
В 1990 году Магнуссон опубликовал основополагающую работу, окончательно исключив значение гибридизации d-орбиталей в связывании гипервалентных соединений элементов второго ряда. Это долгое время было предметом спора и путаницы при описании этих молекул с использованием теории молекулярных орбиталей . Часть путаницы здесь возникает из-за того, что необходимо включать d-функции в базисные наборы, используемые для описания этих соединений (иначе получатся неоправданно высокие энергии и искаженные геометрии), а вклад d-функции в молекулярную волновую функцию велик. Эти факты исторически интерпретировались как означающие, что d-орбитали должны быть вовлечены в связывание. Однако Магнуссон в своей работе приходит к выводу, что участие d-орбиталей не подразумевается в гипервалентности. [5]
Тем не менее, исследование 2013 года показало, что хотя ионная модель Пиментеля лучше всего описывает связывание гипервалентных видов, энергетический вклад расширенной октетной структуры также не равен нулю. В этом современном исследовании теории валентных связей связывания дифторида ксенона было обнаружено, что ионные структуры составляют около 81% общей волновой функции, из которых 70% возникают из ионных структур, использующих только p-орбиталь на ксеноне, в то время как 11% возникают из ионных структур, использующих гибрид на ксеноне. Вклад формально гипервалентной структуры, использующей орбиталь sp 3 d-гибридизации на ксеноне, составляет 11% волновой функции, а бирадикальный вклад составляет оставшиеся 8%. Вклад sp 3 d в размере 11% приводит к чистой стабилизации молекулы на 7,2 ккал (30 кДж) моль −1 , [6] незначительная, но значительная доля полной энергии общей энергии связи (64 ккал (270 кДж) моль −1 ). [7] Другие исследования также обнаружили незначительные, но не пренебрежимо малые энергетические вклады от расширенных октетных структур в SF 6 (17%) и XeF 6 (14%). [8]
Несмотря на отсутствие химического реализма, ИЮПАК рекомендует рисовать расширенные октетные структуры для функциональных групп, таких как сульфоны и фосфораны , чтобы избежать рисования большого количества формальных зарядов или частичных одинарных связей. [9]
Особый тип гипервалентных молекул — гипервалентные гидриды. Большинство известных гипервалентных молекул содержат заместители, более электроотрицательные, чем их центральные атомы. [10] [11] Гипервалентные гидриды представляют особый интерес, поскольку водород обычно менее электроотрицателен, чем центральный атом. Ряд вычислительных исследований был проведен на халькогенгидридах [11] [12] [13] [14] [15] [16] и пниктогенгидридах . [17] [18] [19] [20] [21] Недавно новое вычислительное исследование показало, что большинство гипервалентных галогенгидридов XH n могут существовать. Предполагается, что IH 3 и IH 5 достаточно стабильны, чтобы их можно было наблюдать или, возможно, даже изолировать. [22]
И термин, и концепция гипервалентности по-прежнему подвергаются критике. В 1984 году в ответ на эту общую полемику Пауль фон Раге Шлейер предложил заменить «гипервалентность» на использование термина «гиперкоординация» , поскольку этот термин не подразумевает никакого способа химической связи, и таким образом можно было бы вообще избежать вопроса. [3]
Сама концепция подверглась критике со стороны Рональда Джиллеспи , который на основе анализа функций локализации электронов в 2002 году написал, что «поскольку нет принципиальной разницы между связями в гипервалентных и негипервалентных (октет Льюиса) молекулах, нет причин продолжать использовать термин «гипервалентный»» [23] .
Для гиперкоординированных молекул с электроотрицательными лигандами, такими как PF 5 , было показано, что лиганды могут оттянуть достаточно электронной плотности от центрального атома, так что его чистое содержание снова составит 8 электронов или меньше. Соответствует этой альтернативной точке зрения вывод о том, что гиперкоординированные молекулы на основе фторированных лигандов, например PF 5 , не имеют гидридных аналогов, например фосфорана (PH 5 ), который неизвестен.
Ионная модель хорошо выдерживает термохимические расчеты. Она предсказывает благоприятное экзотермическое образование PF+
4Ф−
из трифторида фосфора PF 3 и фтора F 2 , тогда как аналогичная реакция с образованием PH+
4ЧАС−
не благоприятно. [24]
Дюррант предложил альтернативное определение гипервалентности, основанное на анализе карт атомного заряда, полученных из атомов в теории молекул . [25] Этот подход определяет параметр, называемый эквивалентом валентного электрона, γ, как «формальное общее количество электронов на данном атоме, полученное любой комбинацией действительных ионных и ковалентных резонансных форм, которая воспроизводит наблюдаемое распределение заряда». Для любого конкретного атома X, если значение γ(X) больше 8, этот атом является гипервалентным. Используя это альтернативное определение, многие виды, такие как PCl 5 , SO2−
4, и XeF 4 , которые являются гипервалентными по определению Мушера, переклассифицируются как гиперкоординированные, но не гипервалентные, из-за сильной ионной связи, которая оттягивает электроны от центрального атома. С другой стороны, некоторые соединения, которые обычно записываются с ионными связями, чтобы соответствовать правилу октета, такие как озон O 3 , закись азота NNO и триметиламин N-оксид (CH
3)
3NO , как оказалось, действительно гипервалентны. Примеры γ-расчетов для фосфата PO3−
4(γ(P) = 2,6, негипервалентный) и ортонитрат NO3−
4(γ(N) = 8,5, гипервалентный) показаны ниже.

Ранние рассмотрения геометрии гипервалентных молекул вернули знакомые расположения, которые хорошо объяснялись моделью VSEPR для атомных связей. Соответственно, молекулы типа AB 5 и AB 6 обладали бы тригональной бипирамидальной и октаэдрической геометрией соответственно. Однако для того, чтобы учесть наблюдаемые углы связей, длины связей и кажущееся нарушение правила октета Льюиса , было предложено несколько альтернативных моделей.
В 1950-х годах для объяснения молекулярной архитектуры была предложена расширенная валентная оболочка гипервалентной связи, где центральный атом пента- и гексакоординированных молекул будет использовать d AO в дополнение к s и p AO. Однако прогресс в изучении ab initio расчетов показал, что вклад d-орбиталей в гипервалентную связь слишком мал, чтобы описать свойства связи, и это описание теперь считается гораздо менее важным. [5] Было показано, что в случае гексакоординированного SF 6 d-орбитали не участвуют в образовании связи SF, но перенос заряда между атомами серы и фтора и соответствующие резонансные структуры смогли объяснить гипервалентность (см. ниже).
Дополнительные модификации правила октета были предприняты для включения ионных характеристик в гипервалентную связь. В качестве одной из этих модификаций в 1951 году была предложена концепция 3-центровой 4-электронной (3c-4e) связи , которая описывала гипервалентную связь с качественной молекулярной орбиталью . Связь 3c-4e описывается как три молекулярные орбитали, заданные комбинацией ap атомной орбитали на центральном атоме и атомной орбитали от каждого из двух лигандов на противоположных сторонах центрального атома. Только одна из двух пар электронов занимает молекулярную орбиталь, которая включает связывание с центральным атомом, вторая пара является несвязывающей и занимает молекулярную орбиталь, состоящую только из атомных орбиталей от двух лигандов. Эту модель, в которой сохраняется правило октета, также отстаивал Мушер. [3]

Полное описание гипервалентных молекул возникает из рассмотрения теории молекулярных орбиталей с помощью квантово-механических методов. LCAO , например, в гексафториде серы, беря базисный набор из одной 3s-орбитали серы, трех 3p-орбиталей серы и шести октаэдрических геометрических симметрийно-адаптированных линейных комбинаций (SALC) орбиталей фтора, получают в общей сложности десять молекулярных орбиталей (четыре полностью занятых связывающих МО с самой низкой энергией, две полностью занятых несвязывающих МО с промежуточной энергией и четыре вакантных антисвязывающих МО с самой высокой энергией), предоставляя место для всех 12 валентных электронов. Это стабильная конфигурация только для молекул S X 6, содержащих электроотрицательные атомы лиганда, такие как фтор, что объясняет, почему SH 6 не является стабильной молекулой. В модели связывания две несвязывающие МО (1e g ) локализованы одинаково на всех шести атомах фтора.
Для гипервалентных соединений, в которых лиганды более электроотрицательны , чем центральный, гипервалентный атом, резонансные структуры могут быть нарисованы с не более чем четырьмя ковалентными электронными парными связями и дополнены ионными связями, чтобы соответствовать правилу октета. Например, в пентафториде фосфора (PF 5 ) можно сгенерировать 5 резонансных структур, каждая с четырьмя ковалентными связями и одной ионной связью с большим весом в структурах, помещая ионный характер в аксиальные связи, таким образом удовлетворяя правилу октета и объясняя как наблюдаемую тригонально-бипирамидальную молекулярную геометрию , так и тот факт, что длина аксиальной связи (158 пм) длиннее экваториальной (154 пм). [26]
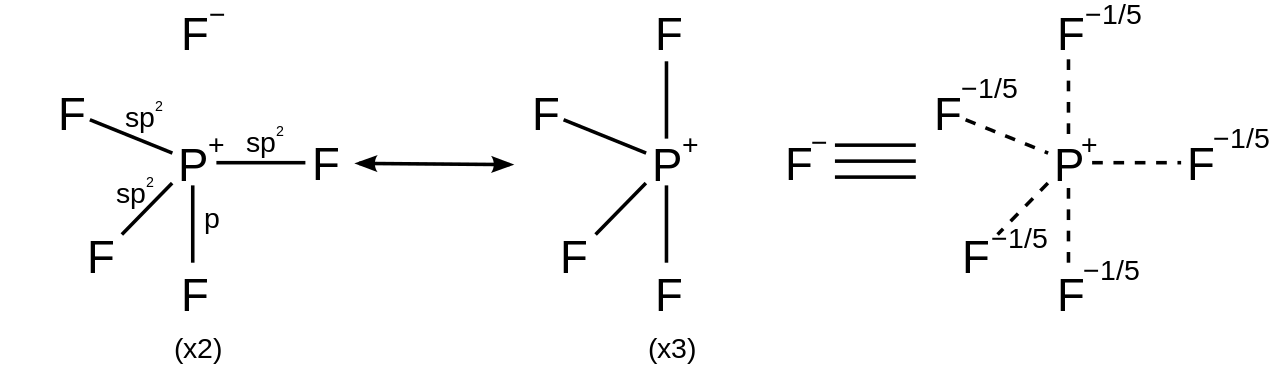
Для гексакоординированной молекулы, такой как гексафторид серы , каждая из шести связей имеет одинаковую длину. Описанное выше обоснование может быть применено для создания 15 резонансных структур, каждая из которых содержит четыре ковалентные связи и две ионные связи, так что ионный характер равномерно распределен по каждой из связей сера-фтор.

Теория спин-связанных валентных связей была применена к диазометану , и полученный орбитальный анализ был интерпретирован в терминах химической структуры, в которой центральный азот имеет пять ковалентных связей;

Это привело авторов к интересному выводу, что «вопреки тому, чему нас учили в студенческие годы, атом азота действительно образует пять ковалентных связей, и наличие или отсутствие d-орбиталей не имеет никакого отношения к такому положению дел». [27]
Гексакоординированные молекулы фосфора , включающие лиганды азота, кислорода или серы, являются примерами гексакоординации кислоты Льюиса и основания Льюиса. [28] Для двух аналогичных комплексов, показанных ниже, длина связи C–P увеличивается с уменьшением длины связи N–P; прочность связи C–P уменьшается с увеличением прочности взаимодействия N–P кислоты Льюиса и основания Льюиса.

Эта тенденция также в целом справедлива для пентакоординированных элементов главной группы с одним или несколькими лигандами, содержащими неподеленные пары, включая примеры кремния с пентакоординированным кислородом, показанные ниже.

Связи Si-галоген варьируются от близких к ожидаемому значению Ван-дер-Ваальса в A (слабая связь) до почти ожидаемого значения одинарной ковалентной связи в C (сильная связь). [28]
Корриу и его коллеги выполнили раннюю работу, характеризующую реакции, которые, как считалось, протекают через гипервалентное переходное состояние. [29] Измерения скоростей реакции гидролиза четырехвалентных хлорсиланов, инкубированных с каталитическими количествами воды, дали скорость, которая является первым порядком по хлорсилану и вторым порядком по воде. Это указывало на то, что две молекулы воды взаимодействовали с силаном во время гидролиза, и из этого был предложен бинуклеофильный механизм реакции. Затем Корриу и его коллеги измерили скорости гидролиза в присутствии нуклеофильного катализатора HMPT, DMSO или DMF. Было показано, что скорость гидролиза снова была первого порядка по хлорсилану, первого порядка по катализатору и теперь первого порядка по воде. Соответственно, скорости гидролиза также демонстрировали зависимость от величины заряда на кислороде нуклеофила.
В совокупности это привело группу к предложению механизма реакции, в котором есть предварительная скорость-определяющая нуклеофильная атака тетракоординированного силана нуклеофилом (или водой), в которой образуется гипервалентный пентакоординированный силан. За этим следует нуклеофильная атака промежуточного соединения водой на этапе, определяющем скорость, приводящая к гексакоординированным видам, которые быстро разлагаются, давая гидроксисилан.
Гидролиз силана был дополнительно исследован Холмсом и его коллегами [30], в которых тетракоординированный Mes
2СиФ
2(Mes = мезитил ) и пентакоординированный Mes
2СиФ−
3были подвергнуты реакции с двумя эквивалентами воды. После двадцати четырех часов гидролиз тетракоординированного силана практически не наблюдался, в то время как пентакоординированный силан был полностью гидролизован через пятнадцать минут. Кроме того, данные рентгеновской дифракции, собранные для тетраэтиламмониевых солей фторсиланов, показали образование решетки бисилонат водорода, поддерживающей гексакоординированный промежуточный продукт, из которого HF−
2быстро замещается, что приводит к гидроксилированному продукту. Эта реакция и кристаллографические данные подтверждают механизм, предложенный Корриу и др .

Очевидная повышенная реактивность гипервалентных молекул, в отличие от тетравалентных аналогов, также наблюдалась для реакций Гриньяра. Группа Корриу измерила [31] полупериоды реакции Гриньяра с помощью ЯМР для родственных калиевых солей 18-краун-6 различных тетра- и пентакоординированных фторсиланов в присутствии каталитических количеств нуклеофила.
Хотя метод полуреакции неточен, магнитудные различия в скоростях реакций позволили предложить схему реакции, в которой предварительная скорость-определяющая атака четырехвалентного силана нуклеофилом приводит к равновесию между нейтральными тетракоординированными видами и анионным пятивалентным соединением. За этим следует нуклеофильная координация двумя реагентами Гриньяра, как обычно наблюдается, образуя гексакоординированное переходное состояние и давая ожидаемый продукт.

Механистические последствия этого распространяются на гексакоординированные виды кремния, которые, как полагают, активны как переходное состояние в некоторых реакциях. Реакция аллил- или кротил -трифторсиланов с альдегидами и кетонами предшествует только активации фторида, чтобы дать пентакоординированный кремний. Этот промежуточный продукт затем действует как кислота Льюиса , чтобы координироваться с атомом кислорода карбонила. Дальнейшее ослабление связи кремний-углерод, когда кремний становится гексакоординированным, помогает управлять этой реакцией. [32]

Подобная реакционная способность также наблюдалась для других гипервалентных структур, таких как смесь фосфорных соединений, для которых были предложены гексакоординированные переходные состояния. Гидролиз фосфоранов и оксифосфоранов был изучен [33] и показано, что он является вторым порядком в воде. Бельский и др . предложили предопределяющую скорость нуклеофильную атаку водой, приводящую к равновесию между пента- и гексакоординированными видами фосфора, за которым следует перенос протона с участием второй молекулы воды в определяющей скорость стадии раскрытия кольца, приводящей к гидроксилированному продукту.

Алкоголиз пентакоординированных фосфорных соединений, таких как триметоксифосфолен с бензиловым спиртом, также, как постулируется, происходит через похожее октаэдрическое переходное состояние, как при гидролизе, однако без раскрытия кольца. [34]

Из этих экспериментов можно понять, что повышенная реакционная способность, наблюдаемая для гипервалентных молекул, по сравнению с аналогичными негипервалентными соединениями, может быть отнесена к конгруэнтности этих видов гиперкоординированным активированным состояниям, обычно образующимся в ходе реакции.
Повышенная реактивность пентакоординированного кремния не полностью изучена. Корриу и его коллеги предположили, что более электроположительный характер пятивалентного атома кремния может быть причиной его повышенной реактивности. [35] Предварительные расчеты ab initio в некоторой степени подтвердили эту гипотезу, но использовали небольшой базисный набор. [36]
Программное обеспечение для ab initio расчетов, Gaussian 86 , использовалось Дитерсом и его коллегами для сравнения тетракоординированного кремния и фосфора с их пентакоординированными аналогами. Этот ab initio подход используется в качестве дополнения для определения того, почему реакционная способность улучшается в нуклеофильных реакциях с пентакоординированными соединениями. Для кремния использовался базисный набор 6-31+G* из-за его пентакоординированного анионного характера, а для фосфора — базисный набор 6-31G* . [36]
Пентакоординированные соединения теоретически должны быть менее электрофильными, чем тетракоординированные аналоги из-за стерических препятствий и большей электронной плотности от лигандов, но экспериментально показывают большую реакционную способность с нуклеофилами, чем их тетракоординированные аналоги. Расширенные расчеты ab initio были выполнены для серий тетракоординированных и пентакоординированных видов, чтобы глубже понять это явление реакционной способности. Каждая серия различалась по степени фторирования. Длины связей и плотности зарядов показаны как функции того, сколько гидридных лигандов находится на центральных атомах. Для каждого нового гидрида имеется на один фторид меньше. [36]
Для длин связей кремния и фосфора, плотностей зарядов и перекрытия связей Малликена популяции были рассчитаны для тетра- и пентакоординированных видов с помощью этого подхода ab initio. [36] Добавление фторид-иона к тетракоординированному кремнию показывает общее среднее увеличение заряда электрона на 0,1, что считается незначительным. В целом, длины связей в тригонально-бипирамидальных пентакоординированных видах длиннее, чем в тетракоординированных аналогах. Связи Si-F и Si-H увеличиваются в длине при пентакоординации, и связанные с этим эффекты наблюдаются в фосфорных видах, но в меньшей степени. Причиной большей величины изменения длины связи для кремниевых видов по сравнению с фосфорными видами является увеличенный эффективный ядерный заряд у фосфора. Поэтому делается вывод, что кремний более слабо связан со своими лигандами.
Кроме того, Дитерс и коллеги [36] показывают обратную корреляцию между длиной связи и перекрытием связей для всех серий. Пентакоординированные виды считаются более реакционноспособными из-за их более слабых связей в виде тригонально-бипирамидальных структур.
При расчете энергий для добавления и удаления фторид-иона в различных видах кремния и фосфора было обнаружено несколько тенденций. В частности, тетракоординированные виды имеют гораздо более высокие энергетические потребности для удаления лиганда, чем пентакоординированные виды. Кроме того, виды кремния имеют более низкие энергетические потребности для удаления лиганда, чем виды фосфора, что является показателем более слабых связей в кремнии.
{{cite book}}: CS1 maint: несколько имен: список авторов ( ссылка ){{cite journal}}: Отсутствует или пусто |title=( помощь )