Программа Управления по контролю за продуктами и лекарствами США по исследованию новых лекарственных препаратов ( IND ) — это способ, с помощью которого фармацевтическая компания получает разрешение на начало клинических испытаний на людях и на отправку экспериментального препарата через границы штатов (обычно клиническим исследователям) до одобрения заявки на маркетинг препарата. Правила в основном изложены в 21 CFR 312. Аналогичные процедуры применяются в Европейском союзе, Японии и Канаде в связи с усилиями по гармонизации регулирования, проводимыми Международным советом по гармонизации . [1]
Типы
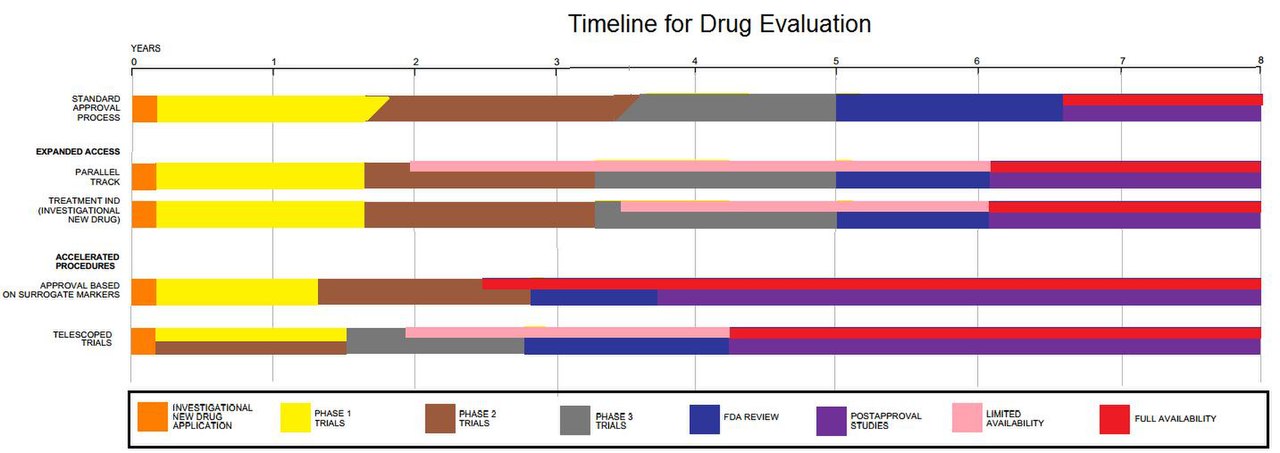
Сроки оценки препарата
Исследовательские или исследовательские IND — это некоммерческие IND, поданные исследователями для изучения неодобренного препарата или изучения одобренного препарата по новому показанию или на новой группе пациентов.
IND для экстренного использования , также называемые сострадательным использованием или IND для одного пациента, подаются для экстренного использования неодобренного препарата, когда клиническая ситуация не позволяет достаточно времени для подачи IND в соответствии с 21 CFR §§ 312.23, 312.24. Они чаще всего используются для опасных для жизни состояний, для которых нет стандартного лечения.
IND на лечение подаются для того, чтобы сделать препарат доступным для лечения серьезных или непосредственно угрожающих жизни состояний до одобрения FDA. Серьезные заболевания или состояния — это инсульт, шизофрения, ревматоидный артрит, остеоартрит, хроническая депрессия, судороги, деменция Альцгеймера, боковой амиотрофический склероз (БАС) и нарколепсия.
Скрининговые IND подаются для нескольких, тесно связанных соединений с целью скрининга предпочтительных соединений или формул. Предпочтительное соединение затем может быть разработано в рамках отдельного IND. Используется для скрининга различных солей, эфиров и других производных лекарственных средств, которые химически различны, но фармакодинамически схожи.
Приложение
Заявку IND можно разделить на следующие категории: [2]
Доклинические испытания состоят из фармакологических и токсикологических исследований на животных для оценки безопасности препарата для тестирования на людях. Также включается любой предыдущий опыт применения препарата на людях (часто зарубежное использование).
Производственная информация включает состав, производителя и стабильность, а также контроль, используемый для производства препарата. Используется для обеспечения того, чтобы компания могла адекватно производить и поставлять последовательные партии препарата.
Информация об исследователе о квалификации клинических исследователей, то есть специалистов (обычно врачей), которые контролируют введение экспериментального препарата субъектам исследования. Используется для оценки того, квалифицированы ли исследователи для выполнения своих обязанностей по клиническим испытаниям.
Протоколы клинических испытаний являются центральным элементом IND. Подробные протоколы для предлагаемых клинических исследований для оценки того, будут ли испытания начальной фазы подвергать субъектов ненужным рискам.
Другие обязательства включают обязательства по получению информированного согласия субъектов исследования, по получению обзора исследования институциональным наблюдательным советом (IRB) и по соблюдению правил, регулирующих новые исследуемые препараты.
Заявка на получение IND также должна включать брошюру исследователя, предназначенную для информирования исследователей о важных фактах об исследуемом препарате, которые им необходимо знать для проведения клинического исследования с наименьшим риском для субъектов или пациентов. [ необходима ссылка ]
После подачи заявки на IND у FDA есть 30 дней, чтобы возразить против IND, иначе он автоматически вступит в силу, и клинические испытания могут начаться. Если FDA обнаружит проблему, оно может наложить клиническую приостановку на IND, запретив начало клинических исследований до тех пор, пока проблема не будет решена, как указано в 21 CFR 312.42 .
На IND должна быть надпись «Внимание: новый препарат — ограничен федеральным законодательством (или законодательством США) для использования в исследовательских целях» в соответствии с 21 CFR 312.6.
FDA проводит программу медицинской марихуаны IND ( программа Compassionate Investigational New Drug ). Она прекратила принимать новых пациентов в 1992 году после того, как органы здравоохранения пришли к выводу, что в ней нет никакой научной ценности, и из-за желания администрации президента Джорджа Буша-старшего «жестко бороться с преступностью и наркотиками». По состоянию на 2011 год четыре пациента продолжают получать каннабис от правительства в рамках этой программы. [3]
^ "E8(R1) Общие положения для клинических исследований" (PDF) . Руководство по эффективности . ICH.
^ Джон С. Макиннес (2011). "Новые применения лекарств". В Shayne C. Gad (ред.). Новые применения лекарств . Энциклопедия фармацевтических наук . doi :10.1002/9780470571224.pse420. ISBN9780470571224.
^ "4 американца получили медицинскую травку от федералов". CBS News . 28 сентября 2011 г.
Внешние ссылки
Центр по оценке и исследованию лекарственных средств для процесса подачи заявок на регистрацию новых исследуемых лекарственных препаратов (IND), Управление по контролю за продуктами питания и лекарственными средствами.
Руководство ICH для промышленности, E6 Надлежащая клиническая практика: консолидированное руководство. НЕРАБОТАЮЩАЯ ССЫЛКА
Тротель, В. М.: Достижение успешной подачи заявления на получение IND в США (1) Журнал по вопросам регулирования. 6: 22–28, январь 1995 г.
Тротель, В. М.: Достижение успешной подачи заявления в IND в США (2) Журнал по вопросам регулирования. 6: 104–108, февраль 1995 г.