Талассемии — это наследственные заболевания крови , которые приводят к аномальному гемоглобину . [7] Симптомы зависят от типа талассемии и могут варьироваться от нулевых до тяжелых. [1] Часто наблюдается легкая или тяжелая анемия (низкий уровень эритроцитов или гемоглобина), поскольку талассемия может влиять на выработку эритроцитов, а также на продолжительность жизни эритроцитов. [1] Симптомы анемии включают чувство усталости и бледность кожи . [1] Другие симптомы талассемии включают проблемы с костями, увеличенную селезенку , желтоватую кожу , легочную гипертензию и темную мочу. [1] У детей может наблюдаться медленный рост. [1] Симптомы и проявления талассемии могут меняться со временем. Более старые термины включали анемию Кули и средиземноморскую анемию для бета-талассемии. Они были заменены терминами «трансфузионно-зависимая талассемия» (ТЗТ) и «нетрансфузионно-зависимая талассемия» (НТЗТ). Пациентам с TDT требуются регулярные переливания, обычно каждые две-пять недель. TDT включают большую бета-талассемию, неделеционную болезнь HbH, выжившую болезнь Hb Барта и тяжелую HbE/бета-талассемию. [8]
Лечение зависит от типа и тяжести. [4] Лечение пациентов с более тяжелым заболеванием часто включает регулярные переливания крови , хелатирование железа и фолиевую кислоту . [4] Хелатирование железа может проводиться с помощью дефероксамина , деферасирокса или деферипрона . [4] [10] Иногда вариантом может быть пересадка костного мозга . [4] Осложнения могут включать перегрузку железом от переливаний с последующими заболеваниями сердца или печени , инфекциями и остеопорозом . [1] Если селезенка становится чрезмерно увеличенной, может потребоваться хирургическое удаление . [1] Пациенты с талассемией, которые плохо реагируют на переливание крови, могут принимать гидроксимочевину или талидомид , а иногда и комбинацию обоих. [11] Гидроксимочевина является единственным препаратом для лечения талассемии, одобренным FDA. Пациенты, принимавшие 10 мг/кг гидроксимочевины каждый день в течение года, имели значительно более высокий уровень гемоглобина, и это было хорошо переносимое лечение для пациентов, которые плохо реагировали на переливание крови. [12] Другие известные индукторы гемоглобина включают талидомид, но он не был испытан в клинических условиях. Сочетание талидомида и гидроксимочевины привело к значительному повышению уровня гемоглобина у пациентов, зависящих и не зависящих от переливания крови [13]
По состоянию на 2015 год талассемия встречается примерно у 280 миллионов человек, причем около 439 000 имеют тяжелую форму заболевания. [14] Она наиболее распространена среди людей греческого , итальянского , ближневосточного , южноазиатского и африканского происхождения. [7] Мужчины и женщины имеют схожие показатели заболеваемости. [ требуется ссылка ] Это привело к 16 800 смертям в 2015 году, что ниже 36 000 смертей в 1990 году. [6] [15] Те, у кого есть легкие степени талассемии, как и те, у кого есть серповидноклеточная анемия , имеют некоторую защиту от малярии , что объясняет, почему серповидноклеточная анемия и талассемия чаще встречаются в регионах мира, где риск малярии выше. [16] По оценкам, 1/3 людей с талассемией имеют «независимую от переливания крови талассемию» и не зависят от регулярных постоянных переливаний крови, чтобы выжить.
Признаки и симптомы
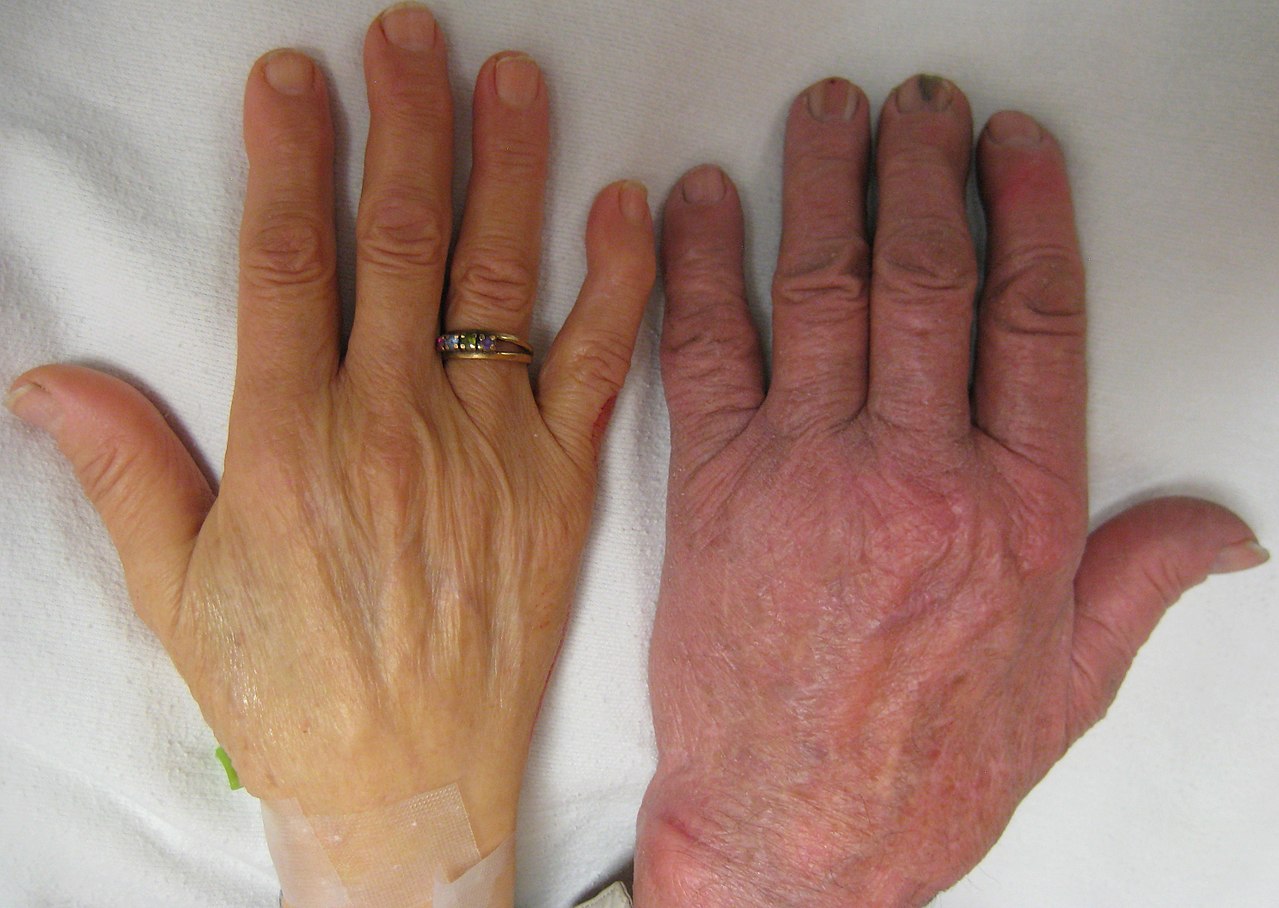
Слева: Рука человека с тяжелой анемией. Справа: Рука человека без анемии.У пациента с талассемией наблюдается увеличение селезенки.
Перегрузка железом : люди с талассемией могут получить перегрузку железом в своих телах, либо из-за самой болезни, либо из-за частых переливаний крови. Слишком много железа может привести к повреждению сердца, печени и эндокринной системы , которая включает железы, вырабатывающие гормоны, регулирующие процессы во всем организме. Повреждение характеризуется чрезмерным отложением железа. Без адекватной терапии хелатирования железа почти все пациенты с бета-талассемией накапливают потенциально смертельные уровни железа. [17]
Инфекция: Люди с талассемией имеют повышенный риск инфекции. Это особенно актуально, если селезенка была удалена. [18]
Деформации костей: Талассемия может привести к расширению костного мозга, что приводит к расширению костей. Это может привести к аномальной структуре костей, особенно лица и черепа. Расширение костного мозга также делает кости тонкими и хрупкими, увеличивая риск переломов. [19]
Увеличенная селезенка : селезенка помогает бороться с инфекцией и фильтрует нежелательный материал, такой как старые или поврежденные клетки крови. Талассемия часто сопровождается разрушением большого количества эритроцитов, и задача удаления этих клеток приводит к увеличению селезенки. Спленомегалия может усугубить анемию и сократить срок службы перелитых эритроцитов. Сильное увеличение селезенки может потребовать ее удаления. [20]
Замедленные темпы роста: анемия может привести к замедлению роста ребенка. Половое созревание также может быть задержано у детей с талассемией. [21]
Проблемы с сердцем: такие заболевания, как застойная сердечная недостаточность и аномальный сердечный ритм, могут быть связаны с тяжелой талассемией. [22]
Структурная биология гемоглобина
Нормальные гемоглобины человека представляют собой тетрамерные белки, состоящие из двух пар цепей глобина, каждая из которых содержит одну альфа-подобную (α-подобную) цепь и одну бета-подобную (β-подобную) цепь. Каждая цепочка глобина связана с железосодержащим гемом. На протяжении всей жизни синтез альфа-подобных и бета-подобных (также называемых не-альфа-подобными) цепей сбалансирован таким образом, что их соотношение относительно постоянно и нет избытка любого типа. [23]
Конкретные альфа- и бета-подобные цепи, входящие в состав Hb, строго регулируются в процессе развития:
Эмбриональные Hb экспрессируются уже на четвертой-шестой неделе эмбриогенеза и исчезают примерно на восьмой неделе беременности, поскольку они заменяются фетальными Hb. [24] [25] Эмбриональные Hb включают:
Hb Gower-1, состоящий из двух ζ-глобинов (дзета-глобинов) и двух ε-глобинов (эпсилон-глобинов) (ζ2ε2)
Hb Gower-2, состоящий из двух альфа-глобинов и двух эпсилон-глобинов (α2ε2)
Hb Portland I, состоящий из двух дзета-глобинов и двух гамма-глобинов (ζ2γ2)
Hb Portland II, состоящий из двух зета-глобинов и двух бета-глобинов (ζ2β2)
Фетальный Hb (Hb F) вырабатывается примерно с восьми недель беременности до рождения и составляет примерно 80 процентов Hb у доношенного новорожденного. Он снижается в течение первых нескольких месяцев жизни и в нормальном состоянии составляет <1 процента от общего Hb к раннему детству и после него. Hb F состоит из двух альфа-глобинов и двух гамма-глобинов (α2γ2). Пациенты с β-талассемией демонстрируют более высокий уровень гамма-глобулина и, следовательно, большую выработку Hb F, чтобы противодействовать дисбалансу из-за невозможности вырабатывать бета-цепи. [26]
Взрослый Hb ( Hb A ) вырабатывается в низких количествах в течение эмбриональной и фетальной жизни и является преобладающим Hb у детей в возрасте от шести месяцев и старше; он составляет 96-97% от общего Hb у людей без гемоглобинопатии. Он состоит из двух альфа-глобинов и двух бета-глобинов (α2β2). [ необходима цитата ]
Hb A2 — это второстепенный взрослый Hb, который обычно составляет около 2,5–3,5% от общего Hb с шестимесячного возраста. Он состоит из двух альфа-глобинов и двух дельта-глобинов (α2δ2). [ необходима цитата ]
Как α-, так и β-талассемии часто наследуются аутосомно - рецессивным способом. Были зарегистрированы случаи доминантно наследуемых α- и β-талассемий, первый из которых был в ирландской семье с двумя делециями 4 и 11 пн в экзоне 3, прерванными вставкой 5 пн в гене β-глобина. Для аутосомно-рецессивных форм заболевания оба родителя должны быть носителями, чтобы ребенок заболел. Если оба родителя несут признак гемоглобинопатии, риск составляет 25% для каждой беременности для больного ребенка. [27]
Гены, вовлеченные в талассемию, контролируют выработку здорового гемоглобина. Гемоглобин связывает кислород в легких и высвобождает его, когда эритроциты достигают периферических тканей, таких как печень. Связывание и высвобождение кислорода гемоглобином необходимы для выживания. [ необходима цитата ]
Эволюция
Наличие одного генетического варианта талассемии может защитить от малярии и, таким образом, может быть преимуществом. [28]
Обычно большая часть гемоглобина взрослого человека ( HbA ) состоит из четырех белковых цепей, двух α- и двух β-глобиновых цепей, организованных в гетеротетрамер . При талассемии у пациентов наблюдаются дефекты в некодирующей области генов α- или β-глобина, что приводит к неэффективному производству нормальных цепей альфа- или бета-глобина, что может привести к неэффективному эритропоэзу, преждевременному разрушению эритроцитов и анемии. [8]
Талассемии классифицируются в зависимости от того, какая цепь молекулы гемоглобина затронута. При α-талассемиях затронуто производство α-глобиновой цепи, тогда как при β-талассемии затронуто производство β-глобиновой цепи. [30]
Цепи β-глобина кодируются одним геном на хромосоме 11 ; цепи α-глобина кодируются двумя тесно связанными генами на хромосоме 16. [ 31] Таким образом, у здорового человека с двумя копиями каждой хромосомы два локуса кодируют цепь β, а четыре локуса кодируют цепь α. Делеция одного из локусов α имеет высокую распространенность у людей африканского или азиатского происхождения, что делает их более склонными к развитию α-талассемии. β-талассемии распространены не только у африканцев , но также у греков и турок . [ требуется цитата ]
Альфа-талассемии
α-талассемии включают гены HBA1 [32] и HBA2 , [33], наследуемые по менделевскому рецессивному типу. Существуют два генных локуса и, таким образом, четыре аллеля. Существуют два генетических локуса для α-глобина, таким образом, четыре аллеля находятся в диплоидных клетках. Два аллеля имеют материнское и два аллеля имеют отцовское происхождение. Тяжесть α-талассемий коррелирует с количеством пораженных α-глобинов; аллели: чем больше, тем тяжелее будут проявления заболевания. [34] Альфа-талассемии приводят к снижению продукции альфа-глобина; следовательно, вырабатывается меньше цепей альфа-глобина, что приводит к избытку β-цепей у взрослых и избытку γ-цепей у новорожденных. Избыток β-цепей образует нестабильные тетрамеры (называемые гемоглобином H или HbH из 4 бета-цепей), которые имеют аномальные кривые диссоциации кислорода. Альфа-талассемии часто встречаются у людей из Юго-Восточной Азии, Ближнего Востока, Китая и у лиц африканского происхождения. [35]
Бета-талассемия
Бета-талассемии обусловлены мутациями в гене HBB на хромосоме 11, [36], также наследующимися по аутосомно-рецессивному типу. Тяжесть заболевания зависит от характера мутации и от наличия мутаций в одном или обоих аллелях. Мутировавшие аллели называются β + , когда частичная функция сохраняется (либо белок имеет сниженную функцию, либо он функционирует нормально, но вырабатывается в сниженном количестве) или β o , когда не вырабатывается функционирующий белок. Положение обоих аллелей определяет клиническую картину:
Большая β-талассемия ( средиземноморская анемия или анемия Кули ) вызывается генотипом β o /β o . Функциональные β-цепи не производятся, и, таким образом, не может быть собран гемоглобин А. Это самая тяжелая форма β-талассемии;
β-талассемия промежуточная вызывается генотипом β + /β o или β + /β + . При этой форме вырабатывается некоторое количество гемоглобина А;
Малая β-талассемия вызывается генотипом β/β o или β/β + . Только один из двух аллелей β-глобина содержит мутацию, поэтому производство β-цепи не сильно нарушено, и пациенты могут быть относительно бессимптомными.
Бета-талассемия чаще всего встречается у людей средиземноморского происхождения. В меньшей степени могут быть затронуты китайцы, другие азиаты и афроамериканцы. [35]
Дельта-талассемия
Помимо альфа- и бета-цепей, присутствующих в гемоглобине, около 3% взрослого гемоглобина состоит из альфа- и дельта-цепей. Как и при бета-талассемии, могут возникнуть мутации, которые влияют на способность этого гена производить дельта-цепи. [37] [38]
Комбинированные гемоглобинопатии
Талассемия может сосуществовать с другими гемоглобинопатиями . Наиболее распространенными из них являются:
Гемоглобин C /талассемия: распространено среди средиземноморских и африканских популяций, гемоглобин C/β o талассемия вызывает умеренно тяжелую гемолитическую анемию со спленомегалией; гемоглобин C/β + талассемия вызывает более легкое заболевание. [ необходима ссылка ]
1) Гипогонадизм: перегрузка железом гонадотропных клеток гипофиза вызывает снижение секреции гонадотропинов, что приводит к задержке, замедлению или остановке полового созревания.
2) Гипопаратиреоз: хроническая анемия вызывает нарушение кроветворения, что приводит к резорбции костей, что снижает секрецию паращитовидных желез.
3) Надпочечниковая недостаточность: снижение роста лобковых и подмышечных волос у подростков с ТМ. Это происходит из-за избыточного отложения железа, что вызывает дисфункцию надпочечников.
4) Гипотиреоз: увеличение веса и замедление роста у пациентов-подростков. Вторичный гипотиреоз встречается редко, большинство страдает первичным гипотиреозом.
[85]
Управление
Учитывая диапазон тяжести, некоторым людям не требуется никакого лечения (тем, у кого нет симптомов), в то время как некоторым людям для выживания требуются регулярные переливания крови. [43] Людям с тяжелой формой талассемии требуется медицинское лечение, и основным методом лечения обычно является переливание эритроцитарной массы.
Переливание эритроцитов
Переливание крови является основным методом лечения для продления жизни. [44] Необходимый подход и частота варьируются у каждого человека в зависимости от тяжести, возраста, задержки роста, наличия экстрамедуллярного эритропоэза (в педиатрии), беременности и состояния сердца. Переливание крови сопряжено с рисками, включая ухудшение перегрузки железом, риск инфекций, риск образования антител эритроцитов, повышенный риск развития реакций гиперчувствительности и риск воспаления желчного пузыря ( холецистита ). [43]
Многократное переливание крови может привести к перегрузке железом. Перегрузку железом, связанную с талассемией, можно лечить с помощью хелатной терапии с помощью препаратов дефероксамин , деферипрон или деферазирокс . [45] [46] [47] Эти методы лечения привели к увеличению продолжительности жизни у людей с большой талассемией. [45] Дефероксамин эффективен только в виде ежедневной инъекции, что осложняет его долгосрочное применение. Однако он недорог и безопасен. К побочным эффектам относятся первичные кожные реакции вокруг места инъекции и потеря слуха . [45] Деферазирокс и деферипрон являются пероральными препаратами, распространенными побочными эффектами которых являются тошнота, рвота и диарея. При сравнении эффективности нет никаких доказательств того, что деферазирокс или деферипрон лучше, однако долгосрочное сравнение не проводилось. [47] Деферазирокс не эффективен для всех пациентов и может не подходить для тех, у кого есть серьезные проблемы с сердцем, связанные с перегрузкой железом, в то время как деферипрон, по-видимому, является наиболее эффективным средством, когда задействовано сердце. Кроме того, стоимость деферазирокса также значительна. [45] Сочетание препаратов блокаторов кальциевых каналов с терапией хелированием железа изучается, однако, преимущества не ясны из проведенных клинических испытаний. [48]
Трансплантация костного мозга может дать возможность излечения молодым людям, у которых есть HLA -совместимый донор. [50] Показатели успеха достигли 80–90%. [50] Смертность от процедуры составляет около 3%. [51] Не существует рандомизированных контролируемых исследований, которые бы проверяли безопасность и эффективность трансплантации костного мозга от неидентичного донора у лиц с β-талассемией, зависящих от переливания крови. [52]
Реакция «трансплантат против хозяина» (РТПХ) является одним из значимых побочных эффектов трансплантации костного мозга. Необходимы дальнейшие исследования, чтобы оценить, можно ли использовать мезенхимальные стромальные клетки для профилактики или лечения РТПХ. [53]
Если у человека нет совместимого донора по HLA, можно попытаться провести трансплантацию костного мозга от гаплоидентичной матери ребенку (несовместимый донор). В исследовании 31 человека выживаемость без талассемии составила 70%, отторжение 23% и смертность 7%. Наиболее положительные результаты, как правило, наблюдаются у очень молодых людей. [54]
генная терапия
Betibeglogene autotemcel , продаваемый под торговой маркой Zynteglo, представляет собой генную терапию для лечения бета-талассемии. [55] Он был одобрен для медицинского применения в Европейском союзе в мае 2019 года, [56] и в Соединенных Штатах в августе 2022 года. [57] [58] Betibeglogene autotemcel показан для лечения людей в возрасте двенадцати лет и старше с бета-талассемией, зависящей от переливания крови, у которых нет генотипа β0/β0, для которых подходит трансплантация гемопоэтических стволовых клеток (ГСК), но отсутствует родственный донор ГСК, соответствующий человеческому лейкоцитарному антигену (HLA).
Процедура включает сбор гемопоэтических стволовых клеток (ГСК) из крови пострадавшего человека. Затем в ГСК добавляют ген бета-глобина с помощью лентивирусного вектора . После разрушения костного мозга пострадавшего человека дозой химиотерапии (режим миелоаблативного кондиционирования) измененные ГСК вливаются обратно пострадавшему человеку, где они приживаются в костном мозге, где они размножаются. Это потенциально приводит к прогрессивному увеличению синтеза гемоглобина А2 во всех последующих развивающихся эритроцитах, что приводит к разрешению анемии. [59]
Лечение было одобрено в Соединенном Королевстве для лечения бета-талассемии, зависящей от переливания крови, в ноябре 2023 года [61] [62] [63] и в Соединенных Штатах в январе 2024 года. [64] [65] [66]
Генная терапия производится из собственных стволовых клеток крови реципиента, которые модифицируются и возвращаются в виде однократной инфузии одной дозы в рамках трансплантации гемопоэтических стволовых клеток . Перед лечением собираются собственные стволовые клетки реципиента, а затем реципиент должен пройти миелоаблативное кондиционирование (высокодозную химиотерапию), процесс, который удаляет клетки из костного мозга, чтобы их можно было заменить модифицированными клетками в exagamglogene autotemcel . Модифицированные стволовые клетки крови трансплантируются обратно реципиенту, где они приживаются в костном мозге и увеличивают выработку фетального гемоглобина (HbF), типа гемоглобина, который облегчает доставку кислорода.
Другие методы лечения
Лечение гидроксимочевиной с целью реактивации гамма-генов для производства HbF не имеет никаких высококачественных доказательств, подтверждающих его эффективность. [67]
Нет никаких доказательств из рандомизированных контролируемых испытаний в поддержку добавления цинка для людей с талассемией. [68] Компьютерные программы или мобильные приложения были предложены в качестве инструментов, помогающих людям контролировать талассемию и следовать их терапии, включая терапию хелированием железа. Эффективность этих приложений не была хорошо исследована. [69]
Люди с талассемией подвержены более высокому риску остеопороза. [70] Варианты лечения включают бисфосфонаты и иногда добавление гормональной терапии. Были предложены другие методы лечения, включая кальцитонин, цинк, гидроксимочевину и добавки кальция. Эффективность бисфосфонатов и цинка не ясна, и требуются дальнейшие исследования. [70]
Стоматологическая помощь
Помощь людям с талассемией в стоматологической помощи и лечении стоматологических проблем может быть сложной из-за основного заболевания. Перегрузка железом из-за переливания крови может привести к отложению железа в зубах и изменению цвета, а также существует повышенный риск инфекции. [71] Варианты лечения для людей с талассемией должны быть изменены, чтобы гарантировать, что потребности человека учитываются, и раннее выявление и раннее управление любыми проблемами настоятельно рекомендуется, чтобы снизить риск того, что человеку потребуется более сложное стоматологическое лечение. [71] Доказательства, подтверждающие эффективность стоматологического лечения для людей с талассемией, слабы, и необходимы более качественные клинические испытания. [71]
Легкая талассемия
Людям с признаками талассемии не требуется медицинская или последующая помощь после постановки первоначального диагноза. [44] Людей с признаками β-талассемии следует предупредить, что их состояние может быть ошибочно диагностировано как более распространенная железодефицитная анемия . Им следует избегать регулярного приема добавок железа , но дефицит железа может развиться во время беременности или из-за хронического кровотечения. [72] Генетическое консультирование показано всем лицам с генетическими нарушениями, особенно когда семья подвержена риску тяжелой формы заболевания, которую можно предотвратить. [73]
Профилактика
Американский колледж акушеров и гинекологов рекомендует всем женщинам, планирующим беременность, пройти обследование на наличие талассемии. [74] Генетическое консультирование и генетическое тестирование рекомендуются семьям, в которых есть признаки талассемии. [27] Понимание генетического риска, в идеале до того, как семья заведет ребенка, позволит семьям лучше понять это состояние и принять обоснованное решение, которое будет наилучшим для их семьи. [27]
На Кипре существует политика скрининга для снижения уровня талассемии, которая с момента внедрения программы в 1970-х годах (включая также пренатальный скрининг и аборты) сократила количество детей, рожденных с этим заболеванием, с одного на каждые 158 родов почти до нуля. [75] В Греции также есть программа скрининга для выявления людей, являющихся носителями. [76]
В Иране в качестве предсвадебного скрининга сначала проверяют индексы эритроцитов у мужчины. Если у него микроцитоз ( средний гемоглобин клеток < 27 пг или средний объем эритроцитов < 80 фл), то тестируют женщину. Если у обоих микроцитоз, измеряют концентрацию гемоглобина А2 . Если у обоих концентрация выше 3,5% (диагностический признак талассемии), их направляют в местный назначенный медицинский пункт для генетического консультирования . [77]
В Индии [78] как правительственные, так и неправительственные организации организуют широкомасштабные кампании по повышению осведомленности в целях поощрения добровольного добрачного скрининга, при этом браки между носителями настоятельно не приветствуются.
Эпидемиология
Бета-форма талассемии особенно распространена среди средиземноморских народов, и эта географическая ассоциация ответственна за ее первоначальное название. [79] Талассемия привела к 25 000 смертей в 2013 году по сравнению с 36 000 смертей в 1990 году. [15]
Заболевание также встречается у популяций, проживающих в Африке, Америке и у народа тхару в регионе Терай в Непале и Индии . [81] Считается, что это объясняет гораздо более низкие показатели заболеваемости и смертности от малярии, [82] объясняя историческую способность тхару выживать в районах с сильным заражением малярией, в то время как другие не могли. Талассемии особенно связаны с людьми средиземноморского происхождения, арабами (особенно палестинцами и людьми палестинского происхождения) и азиатами. [83] Предполагаемая распространенность составляет 16% у людей с Кипра , 1% [84] в Таиланде и 3–8% у популяций из Бангладеш , Китая , Индии , Малайзии и Пакистана .
По оценкам, приблизительно 1,5% населения мира (80–90 миллионов человек) являются носителями β-талассемии. [85] Однако точные данные о показателях носительства во многих популяциях отсутствуют, особенно в развивающихся регионах мира, которые, как известно или как ожидается, сильно затронуты. [86] [87] Из-за распространенности заболевания в странах с небольшими знаниями о талассемии, доступ к надлежащему лечению и диагностике может быть затруднен. [88] Хотя в развивающихся странах есть некоторые диагностические и лечебные учреждения, в большинстве случаев они не предоставляются государственными службами и доступны только пациентам, которые могут себе их позволить. В целом, более бедные слои населения имеют доступ только к ограниченным диагностическим учреждениям и переливаниям крови. В некоторых развивающихся странах практически нет учреждений для диагностики или лечения талассемии. [88]
Этимология и синонимы
Слово талассемия ( / θ æ l ɪ ˈ s iː m i ə / ) происходит от греческого thalassa (θάλασσα), «море», [89] и неолатинского -emia (от греческой составной основы - aimia (-αιμία), от haima (αἷμα), «кровь»). [90] Оно было придумано, потому что состояние, называемое «средиземноморской анемией», было впервые описано у людей средиземноморских этнических групп. «Средиземноморская анемия» была переименована в большую талассемию , когда генетика была лучше понята. Слово талассемия было впервые использовано в 1932 году. [79] : 877 [91]
Исследовать
Ссылки
^ abcdefgh «Каковы признаки и симптомы талассемии?». NHLBI . 3 июля 2012 г. Архивировано из оригинала 16 сентября 2016 г. Получено 5 сентября 2016 г.
^ abc "What Causes Thalassemias?". NHLBI . 3 июля 2012 г. Архивировано из оригинала 26 августа 2016 г. Получено 5 сентября 2016 г.
^ ab "Как диагностируется талассемия?". NHLBI . 3 июля 2012 г. Архивировано из оригинала 16 сентября 2016 г. Получено 5 сентября 2016 г.
^ abcde «Как лечат талассемии?». NHLBI . 3 июля 2012 г. Архивировано из оригинала 16 сентября 2016 г. Получено 5 сентября 2016 г.
^ ГББ 2015 Заболеваемость и распространенность травм и заболеваний (8 октября 2016 г.). «Глобальная, региональная и национальная заболеваемость, распространенность и годы, прожитые с инвалидностью, для 310 заболеваний и травм, 1990–2015 гг.: систематический анализ для исследования глобального бремени болезней 2015 г.». Lancet . 388 (10053): 1545–1602. doi :10.1016/S0140-6736(16)31678-6. PMC 5055577 . PMID 27733282.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ ab GBD 2015 Смертность и причины смерти (8 октября 2016 г.). «Глобальная, региональная и национальная ожидаемая продолжительность жизни, смертность от всех причин и смертность по конкретным причинам для 249 причин смерти, 1980–2015 гг.: систематический анализ для исследования глобального бремени болезней 2015 г.». Lancet . 388 (10053): 1459–1544. doi :10.1016/s0140-6736(16)31012-1. PMC 5388903 . PMID 27733281.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ abc "Что такое талассемии?". NHLBI . 3 июля 2012 г. Архивировано из оригинала 26 августа 2016 г. Получено 5 сентября 2016 г.
^ ab Baird DC, Batten SH, Sparks SK. Альфа- и бета-талассемия: быстрый обзор доказательств. Am Fam Physician. 2022 1 марта;105(3):272-280. PMID 35289581.
^ «Как предотвратить талассемию?». NHLBI . 3 июля 2012 г. Архивировано из оригинала 16 сентября 2016 г. Получено 5 сентября 2016 г.
^ "Хелатирование железа" . Получено 15 июля 2020 г.
^ Шах, Сандип; Шет, Радхика; Шах, Камлеш; Патель, Киннари (февраль 2020 г.). «Безопасность и эффективность комбинации талидомида и гидроксимочевины при промежуточной и большой β-талассемии: ретроспективное пилотное исследование». British Journal of Haematology . 188 (3): e18–e21. doi : 10.1111/bjh.16272 . ISSN 0007-1048. PMID 31710694. S2CID 207940189.
^ Keikhaei, Bijan (2015). «Клинические и гематологические эффекты гидроксимочевины у пациентов с β-талассемией промежуточной». Журнал клинических и диагностических исследований . 9 (10): OM01-3. doi :10.7860/JCDR/2015/14807.6660. PMC 4625280. PMID 26557561 .
^ Мазера, Николетта; Тавеккья, Луиза; Капра, Мариетта; Каццанига, Джованни; Вимеркати, Кьяра; Поцци, Лорена; Бионди, Андреа; Мазера, Джузеппе (2010). «Оптимальный ответ на талидомид у пациента с большой талассемией, резистентного к традиционной терапии». Переливание крови . 8 (1): 63–5. дои : 10.2450/2009.0102-09. ISSN 1723-2007. ПМК 2809513 . ПМИД 20104280.
^ Исследование глобального бремени болезней 2013 г. (22 августа 2015 г.). «Глобальная, региональная и национальная заболеваемость, распространенность и годы, прожитые с инвалидностью, для 301 острого и хронического заболевания и травм в 188 странах, 1990–2013 гг.: систематический анализ для исследования глобального бремени болезней 2013 г.». Lancet . 386 (9995): 743–800. doi :10.1016/s0140-6736(15)60692-4. PMC 4561509 . PMID 26063472.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ ab GBD 2013 Смертность и причины смерти (17 декабря 2014 г.). «Глобальная, региональная и национальная возрастно-половая специфическая смертность от всех причин и причинно-специфических причин по 240 причинам смерти, 1990–2013 гг.: систематический анализ для исследования глобального бремени болезней 2013 г.». The Lancet . 385 (9963): 117–71. doi :10.1016/S0140-6736(14)61682-2. hdl :11655/15525. PMC 4340604 . PMID 25530442.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ Weatherall, DJ (2015). «Талассемии: нарушения синтеза глобина». Williams Hematology (9-е изд.). McGraw Hill Professional. стр. 725. ISBN9780071833011.
^ Cianciulli P (октябрь 2008 г.). «Лечение перегрузки железом при талассемии». Pediatr Endocrinol Rev. 6 ( Suppl 1): 208–13. PMID 19337180.
^ "Талассемия – Симптомы и причины". Mayo Clinic . Архивировано из оригинала 20 ноября 2016 года . Получено 4 апреля 2017 года .
^ Vogiatzi, Maria G; Macklin, Eric A; Fung, Ellen B; Cheung, Angela M; Vichinsky, Elliot; Olivieri, Nancy; Kirby, Melanie; Kwiatkowski, Janet L; Cunningham, Melody; Holm, Ingrid A; Lane, Joseph; Schneider, Robert; Fleisher, Martin; Grady, Robert W; Peterson, Charles C; Giardina, Patricia J (март 2009 г.). «Заболевания костей при талассемии: частая и все еще нерешенная проблема». Journal of Bone and Mineral Research . 24 (3): 543–557. doi :10.1359/jbmr.080505. ISSN 0884-0431. PMC 3276604. PMID 18505376 .
^ "Симптомы и причины – Увеличенная селезенка (спленомегалия) – Клиника Майо". www.mayoclinic.org . Архивировано из оригинала 19 ноября 2016 года . Получено 2 февраля 2017 года .
^ Солиман, Ашраф Т; Калра, Санджай; Де Санктис, Винченцо (1 ноября 2014 г.). «Анемия и рост». Индийский журнал эндокринологии и метаболизма . 18 (7): S1–5. doi : 10.4103/2230-8210.145038 . PMC 4266864. PMID 25538873 .
^ "Осложнения талассемии". Талассемия . Открытое издание. Архивировано из оригинала 3 октября 2011 г. Получено 27 сентября 2011 г.
^ Weatherall DJ. Новая генетика и клиническая практика, Oxford University Press, Оксфорд, 1991.
^ Хейсман Т. Х. Структура и функция нормальных и аномальных гемоглобинов. В: Baillière's Clinical Hematology, Higgs DR, Weatherall DJ (Eds), WB Saunders, Лондон, 1993. стр. 1.
^ Natarajan K, Townes TM, Kutlar A. Нарушения структуры гемоглобина: серповидноклеточная анемия и связанные с ней аномалии. В: Williams Hematology, 8-е изд., Kaushansky K, Lichtman MA, Beutler E, et al. (редакторы), McGraw-Hill, 2010. стр. 48.
^ Вай Фенг Лим, Логесваран Мунианди, Элизабет Джордж, Джамила Сатхар, Лай Куан Тэ и Мэй И Лай (2015) HbF в HbE/β-талассемии: клиническая и лабораторная корреляция, Гематология, 20:6, 349-353, DOI: 10.1179/1607845414Y.0000000203
^ abc Хуссейн, Норита; Хеннеман, Лидевий; Кай, Джо; Куреши, Надим (11 октября 2021 г.). Группа по муковисцидозу и генетическим расстройствам Кокрейна (ред.). «Оценка риска до зачатия при талассемии, серповидноклеточной анемии, муковисцидозе и болезни Тея-Сакса». База данных систематических обзоров Кокрейна . 2021 (10): CD010849. doi : 10.1002 /14651858.CD010849.pub4. PMC 8504980. PMID 34634131.
^ Вамбуа С.; Мванги, Табита В.; Корток, Моисей; Уйога, Софи М.; Мачария, Алекс В.; Мвачаро, Джедида К.; Уэзеролл, Дэвид Дж.; Сноу, Роберт В.; Марш, Кевин; Уильямс, Томас Н. (май 2006 г.). «Влияние α +-талассемии на заболеваемость малярией и другими заболеваниями у детей, живущих на побережье Кении». PLOS Medicine . 3 (5): e158. doi : 10.1371/journal.pmed.0030158 . PMC 1435778. PMID 16605300 .
^ Тассиопулос С; Дефтереос, Спирос; Константинопулос, Костас; Фармакис, Димитрис; Цирони, Мария; Кириакидис, Михалис; Эссоп, Афанассиос (2005). «Дает ли гетерозиготная бета-талассемия защиту от ишемической болезни сердца?». Анналы Нью-Йоркской академии наук . 1054 (1): 467–70. Бибкод : 2005NYASA1054..467T. дои : 10.1196/анналы.1345.068. PMID 16339699. S2CID 71993591.
^ Герберт Л. Манси, младший; Кэмпбелл, Джеймс С. (15 августа 2009 г.). «Альфа и бета-талассемия». American Family Physician . 80 (4): 339–344. PMID 19678601.
^ ab Galanello, Renzo; Cao, Antonio (5 января 2011 г.). «Альфа-талассемия». Genetics in Medicine . 13 (2): 83–88. doi : 10.1097/GIM.0b013e3181fcb468 . ISSN 1098-3600. PMID 21381239.
^ ab "Основы анемии". WebMD . Получено 9 мая 2019 г.
^ abcd Neufeld, EJ (2010). «Обновление по хелаторам железа при талассемии». Гематология . 2010 : 451–5. doi : 10.1182/asheducation-2010.1.451 . PMID 21239834.
^ Фишер, Шейла А.; Бранскилл, Сьюзан Дж.; Дори, Кэролин; Чоудхури, Онима; Гудинг, Сара; Робертс, Дэвид Дж. (21 августа 2013 г.). «Пероральный деферипрон для хелатирования железа у людей с талассемией». База данных систематических обзоров Кокрейна (8): CD004839. doi :10.1002/14651858.CD004839.pub3. ISSN 1469-493X. PMID 23966105.
^ ab Bollig, Claudia; Schell, Lisa K.; Rücker, Gerta; Allert, Roman; Motschall, Edith; Niemeyer, Charlotte M.; Bassler, Dirk; Meerpohl, Joerg J. (15 августа 2017 г.). "Деферазирокс для лечения перегрузки железом у людей с талассемией". База данных систематических обзоров Cochrane . 2017 (8): CD007476. doi :10.1002/14651858.CD007476.pub3. ISSN 1469-493X. PMC 6483623. PMID 28809446 .
^ Садаф, Алина; Хасан, Бабар; Дас, Джай К; Колан, Стивен; Альви, Наджвин (12 июля 2018 г.). Группа по муковисцидозу и генетическим расстройствам Кокрейна (ред.). «Блокаторы кальциевых каналов для профилактики кардиомиопатии из-за перегрузки железом у людей с трансфузионно-зависимой бета-талассемией». База данных систематических обзоров Кокрейна . 2018 (7): CD011626. doi :10.1002/14651858.CD011626.pub2. PMC 6513605. PMID 29998494.
^ Ngim, CF; Lai, NM; Hong, JY; Tan, SL; Ramadas, A; Muthukumarasamy, P; Thong, MK (28 мая 2020 г.). «Терапия гормоном роста для людей с талассемией». База данных систематических обзоров Cochrane . 2020 (5): CD012284. doi :10.1002/14651858.CD012284.pub3. PMC 7387677. PMID 32463488 .
^ ab Газиев, Дж.; Лукарелли, Г. (июнь 2011 г.). «Трансплантация гемопоэтических стволовых клеток при талассемии». Current Stem Cell Research & Therapy . 6 (2): 162–9. doi :10.2174/157488811795495413. PMID 21190532.
^ Sabloff, M; Chandy, M; Wang, Z; Logan, BR; Ghavamzadeh, A; Li, CK; Irfan, SM; Bredeson, CN; et al. (2011). «Трансплантация костного мозга от сиблингов, соответствующих HLA, при большой β-талассемии». Blood . 117 (5): 1745–50. doi :10.1182/blood-2010-09-306829. PMC 3056598 . PMID 21119108.
^ Шарма, Акшай; Джаганнат, Ванита А.; Пури, Латика (21 апреля 2021 г.). «Трансплантация гемопоэтических стволовых клеток для людей с β-талассемией». База данных систематических обзоров Кокрейна . 2021 (4): CD008708. doi :10.1002/14651858.CD008708.pub5. ISSN 1469-493X. PMC 8078520. PMID 33880750 .
^ Фишер, Шейла А.; Катлер, Энтони; Дори, Кэролин; Бранскилл, Сьюзан Дж.; Стэнворт, Саймон Дж.; Наваррете, Кристина; Гердлстоун, Джон (30 января 2019 г.). Группа по гематологическим злокачественным новообразованиям Кокрейна (ред.). «Мезенхимальные стромальные клетки как лечение или профилактика острой или хронической реакции «трансплантат против хозяина» у реципиентов трансплантата гемопоэтических стволовых клеток (ТГСК) с гематологическим заболеванием». База данных систематических обзоров Кокрейна . 1 (1): CD009768. doi :10.1002/14651858.CD009768.pub2. PMC 6353308. PMID 30697701 .
^ Содани, П; Исгро, А; Газиев Дж.; Пачиарони, К; Марциали, М; Симона, доктор медицины; Роведа, А; Де Анджелис, Дж; и др. (2011). «Трансплантация hla-гаплоидентичных стволовых клеток с обеднением Т-клеток молодым пациентам с талассемией». Педиатрические отчеты . 3 (Дополнение 2): e13. doi :10.4081/pr.2011.s2.e13. ПМК 3206538 . ПМИД 22053275.
^ "Zynteglo EPAR". Европейское агентство по лекарственным средствам (EMA) . 25 марта 2019 г. Архивировано из оригинала 16 августа 2019 г. Получено 16 августа 2019 г.Текст был скопирован из этого источника, авторские права на который принадлежат Европейскому агентству по лекарственным средствам. Воспроизведение разрешено при условии указания источника.
^ "Zynteglo". Управление по контролю за продуктами и лекарствами США . 17 августа 2022 г. Архивировано из оригинала 26 августа 2022 г. Получено 26 августа 2022 г.
^ "FDA одобряет первую клеточную генную терапию для лечения взрослых и детей с бета-талассемией, которым требуются регулярные переливания крови". Управление по контролю за продуктами и лекарствами США (FDA) (пресс-релиз). 17 августа 2022 г. Архивировано из оригинала 21 августа 2022 г. Получено 20 августа 2022 г.В данной статье использован текст из этого источника, находящегося в общественном достоянии .
^ Biffi, A (19 апреля 2018 г.). «Генная терапия как метод лечения β-талассемии». The New England Journal of Medicine . 378 (16): 1551–1552. doi :10.1056/NEJMe1802169. PMID 29669229.
^ Stein R (31 октября 2023 г.). «Консультанты FDA не видят препятствий для лечения серповидноклеточной анемии с помощью редактирования генов». NPR . Архивировано из оригинала 4 декабря 2023 г. Получено 4 декабря 2023 г.
^ "MHRA разрешает первую в мире генную терапию, направленную на лечение серповидноклеточной анемии и β-талассемии, зависящей от переливания крови". Агентство по регулированию лекарственных средств и изделий медицинского назначения (MHRA) (пресс-релиз). 16 ноября 2023 г. Архивировано из оригинала 25 ноября 2023 г. Получено 8 декабря 2023 г.
^ Sheridan C (ноябрь 2023 г.). «Первая в мире терапия CRISPR одобрена: кто ее получит?». Nature Biotechnology . 42 (1): 3–4. doi :10.1038/d41587-023-00016-6. PMID 37989785. S2CID 265350318. Архивировано из оригинала 4 декабря 2023 г. Получено 4 декабря 2023 г.
^ «Vertex и CRISPR Therapeutics объявляют об авторизации первой терапии с редактированием генов CRISPR/Cas9, Casgevy (exagamglogene autotemcel), Управлением по контролю за оборотом лекарственных средств и лекарственных препаратов Великобритании (MHRA) для лечения серповидноклеточной анемии и бета-талассемии, зависящей от переливания крови» (пресс-релиз). Vertex Pharmaceuticals. 16 ноября 2023 г. Архивировано из оригинала 22 ноября 2023 г. Получено 9 декабря 2023 г. – через Business Wire.
^ ab "FDA одобряет первые генные терапии для лечения пациентов с серповидноклеточной анемией". Управление по контролю за продуктами и лекарствами США (FDA) . 8 декабря 2023 г. Архивировано из оригинала 8 декабря 2023 г. Получено 8 декабря 2023 г.В данной статье использован текст из этого источника, находящегося в общественном достоянии .
^ «Vertex и CRISPR Therapeutics объявляют об одобрении FDA США препарата Casgevy (exagamglogene autotemcel) для лечения серповидноклеточной анемии» (пресс-релиз). Vertex Pharmaceuticals. 8 декабря 2023 г. Архивировано из оригинала 9 декабря 2023 г. Получено 9 декабря 2023 г. – через Business Wire.
↑ Комиссар, Офис (16 января 2024 г.). "FDA Roundup: January 16, 2024". FDA . Получено 19 января 2024 г. .
^ Ансари, Сакиб Х.; Ласси, Зохра С.; Ховайя, Салима М.; Адил, Сайед Омайр; Шамси, Тахир С. (16 марта 2019 г.). Группа по кистозному фиброзу и генетическим расстройствам Кокрейна (ред.). «Гидроксимочевина (гидроксикарбамид) при трансфузионно-зависимой β-талассемии». База данных систематических обзоров Кокрейна . 3 (3): CD012064. doi : 10.1002 /14651858.CD012064.pub2. PMC 6421980. PMID 30882896.
^ Kye Mon Min Swe (2013). «Добавки цинка для лечения талассемии и серповидноклеточной анемии». База данных систематических обзоров Cochrane . 2013 (6): CD009415. doi :10.1002/14651858.CD009415.pub2. PMC 9964104. PMID 23807756 .
^ Mulimani, Priti; Abas, Adinegara BL; Karanth, Laxminarayan; Colombatti, Raffaella; Kulkarni, Palna (2 августа 2019 г.). Cochrane Mustic Fibrosis and Genetic Disorders Group (ред.). «Лечение стоматологических и ортодонтических осложнений при талассемии». Cochrane Database of Systematic Reviews . 8 (8): CD012969. doi :10.1002/14651858.CD012969.pub2. PMC 6699676. PMID 31425614 .
^ ab Bhardwaj, Amit; Swe, Kye Mon Min; Sinha, Nirmal K.; Osunkwo, Ifeyinwa (10 марта 2016 г.). «Лечение остеопороза у людей с ß-талассемией». База данных систематических обзоров Cochrane . 3 : CD010429. doi :10.1002/14651858.CD010429.pub2. ISSN 1469-493X. PMID 26964506.
^ abc Mulimani, Priti; Abas, Adinegara BL; Karanth, Laxminarayan; Colombatti, Raffaella; Kulkarni, Palna (2 февраля 2023 г.). Cochrane Mustic Fibrosis and Genetic Disorders Group (ред.). "Лечение стоматологических и ортодонтических осложнений при талассемии". Cochrane Database of Systematic Reviews . 2023 (2): CD012969. doi :10.1002/14651858.CD012969.pub3. PMC 9893875. PMID 36732291 .
^ Burdick CO; Ntaios, G.; Rathod, D. (март 2009 г.). «Разделение признака талассемии и дефицита железа путем простого осмотра». Am. J. Clin. Pathol . 131 (3): 444, ответ автора 445. doi : 10.1309/AJCPC09VRAXEASMH . PMID 19228649.
^ "Carrier Screening in the Age of Genomic Medicine – ACOG". www.acog.org . Архивировано из оригинала 25 февраля 2017 г. . Получено 24 февраля 2017 г. .
^ Leung TN; Lau TK; Chung TKh (апрель 2005 г.). «Скрининг талассемии во время беременности». Current Opinion in Obstetrics and Gynecology . 17 (2): 129–34. doi :10.1097/01.gco.0000162180.22984.a3. PMID 15758603. S2CID 41877258.
^ Loukopoulos, D (октябрь 2011 г.). «Гемоглобинопатии в Греции: программа профилактики за последние 35 лет». Индийский журнал медицинских исследований . 134 (4): 572–6. PMC 3237258. PMID 22089622 .
^ Samavat A, Modell B (ноябрь 2004 г.). «Иранская национальная программа скрининга талассемии». BMJ (Clinical Research Ed.) . 329 (7475): 1134–7. doi :10.1136/bmj.329.7475.1134. PMC 527686. PMID 15539666 .
^ Петроу, Мэри (1 января 2010 г.). «Скрининг на бета-талассемию». Indian Journal of Human Genetics . 16 (1): 1–5. doi : 10.4103/0971-6866.64934 . PMC 2927788. PMID 20838484 .[ постоянная мертвая ссылка ]
^ ab Грир, Джон П.; Арбер, Дэниел А.; Глэйдер, Бертил; Лист, Алан Ф.; Минс, младший, Роберт Т.; Параскевас, Фриксос; Роджерс, Джордж М.; Фоерстер, Джон (2013). Клиническая гематология Винтроуба . Wolters Kluwer, Lippincott Williams & Wilkins Health. ISBN9781451172683.
^ Waheed, Fazeela; Fishter, Colleen; Awofeso, AwoNiyi; Stanley, David (июль 2016 г.). «Скрининг носителей бета-талассемии на Мальдивах: восприятие родителей пораженных детей, которые не принимали участия в скрининге, и его последствия». Journal of Community Genetics . 7 (3): 243–253. doi :10.1007/s12687-016-0273-5. PMC 4960032 . PMID 27393346.
^ Модиано, Г.; Морпурго, Г.; Терренато, Л.; Новеллетто, А.; Ди Риенцо, А.; Коломбо, Б.; Пурпура, М.; Мариани, М.; и др. (1991). «Защита от заболеваемости малярией: почти фиксация гена α-талассемии в непальской популяции». Американский журнал генетики человека . 48 (2): 390–7. PMC 1683029. PMID 1990845 .
^ Terrenato, L; Shrestha, S; Dixit, KA; Luzzatto, L; Modiano, G; Morpurgo, G; Arese, P (февраль 1988 г.). «Снижение заболеваемости малярией среди народа тхару по сравнению с симпатрическими популяциями в Непале». Annals of Tropical Medicine and Parasitology . 82 (1): 1–11. doi :10.1080/00034983.1988.11812202. PMID 3041928.
^ Вичинский, Эллиотт П. (1 ноября 2005 г.). «Изменение моделей талассемии во всем мире». Анналы Нью-Йоркской академии наук . 1054 (1): 18–24. Bibcode : 2005NYASA1054...18V. doi : 10.1196/annals.1345.003. ISSN 1749-6632. PMID 16339647. S2CID 26329509.
^ ab Weatherall, David J. (ноябрь 2005 г.). «Основной доклад: проблема талассемии для развивающихся стран». Анналы Нью-Йоркской академии наук . 1054 (1): 11–17. Bibcode : 2005NYASA1054...11W. doi : 10.1196/annals.1345.002. PMID 16339646. S2CID 45770891.
^ Whipple, GH; Bradford, WI (1932). «Расовая или семейная анемия у детей, связанная с фундаментальными нарушениями метаболизма костей и пигмента (Кули-фон Якша)». Американский журнал детских болезней . 44 : 336–365. doi :10.1001/archpedi.1932.01950090074009.