Вычислительная химия — это раздел химии , который использует компьютерное моделирование для решения химических задач. [1] Он использует методы теоретической химии , включенные в компьютерные программы для расчета структур и свойств молекул , групп молекул и твердых тел. [2] Важность этой темы обусловлена тем фактом, что, за исключением некоторых относительно недавних открытий, связанных с молекулярным ионом водорода ( катионом диводорода ), достижение точного квантовомеханического описания химических систем аналитически или в замкнутой форме, не осуществимо. [3] Сложность, присущая проблеме многих тел, усугубляет проблему детального описания квантово-механических систем. [4] Хотя результаты вычислений обычно дополняют информацию, полученную в ходе химических экспериментов , иногда они могут предсказывать ненаблюдаемые химические явления . [5]

Вычислительная химия отличается от теоретической химии , которая предполагает математическое описание химии. Однако вычислительная химия предполагает использование компьютерных программ и дополнительных математических навыков для точного моделирования различных химических задач. В теоретической химии химики, физики и математики разрабатывают алгоритмы и компьютерные программы для прогнозирования атомных и молекулярных свойств и путей реакций химических реакций. Компьютерные химики, напротив, могут просто применять существующие компьютерные программы и методологии для решения конкретных химических вопросов. [7]
Исторически вычислительная химия имела два разных аспекта:
Эти аспекты, наряду с целью вычислительной химии, привели к созданию целого ряда алгоритмов.
Основываясь на основополагающих открытиях и теориях в истории квантовой механики , первые теоретические расчеты в химии были проведены Уолтером Гейтлером и Фрицем Лондоном в 1927 году с использованием теории валентных связей . [9] К книгам, которые оказали влияние на раннее развитие вычислительной квантовой химии, относятся « Введение в квантовую механику – с приложениями к химии» Лайнуса Полинга и Э. Брайта Уилсона 1935 года , [10] « Квантовая химия» Айринга , Уолтера и Кимбалла 1944 года , [ 9] 11] « Элементарная волновая механика» Гейтлера 1945 года – с приложениями к квантовой химии [ 12] и более поздний учебник Коулсона « Валентность » 1952 года, каждый из которых служил основным справочником для химиков в последующие десятилетия. [13]
С развитием эффективных компьютерных технологий в 1940-х годах решение сложных волновых уравнений для сложных атомных систем стало достижимой целью. В начале 1950-х годов были выполнены первые полуэмпирические расчеты атомных орбиталей. Химики-теоретики стали активными пользователями первых цифровых компьютеров. Одно значительное достижение было отмечено статьей Клеменса Рутана в 1951 году в «Обзорах современной физики». [14] [15] Эта статья в основном посвящена подходу «LCAO MO» (линейная комбинация атомных орбиталей, молекулярных орбиталей). В течение многих лет это была вторая по цитируемости статья в этом журнале. [14] [15] Очень подробный отчет о таком использовании в Соединенном Королевстве дан Смитом и Сатклиффом. [16] Первые ab initio расчеты методом Хартри-Фока для двухатомных молекул были выполнены в 1956 году в Массачусетском технологическом институте с использованием базисного набора орбиталей Слейтера . [17] Для двухатомных молекул систематическое исследование с использованием минимального базисного набора и первые расчеты с более крупным базисным набором были опубликованы Рансилом и Несбетом соответственно в 1960 году. [18] Первые многоатомные расчеты с использованием гауссовых орбиталей были выполнены в конце 1950-х годов. . Первые расчеты конфигурационного взаимодействия были выполнены в Кембридже на компьютере EDSAC в 1950-х годах с использованием гауссовских орбиталей Бойсом и его коллегами. [19] К 1971 году, когда была опубликована библиография расчетов ab initio , [20] крупнейшими включенными молекулами были нафталин и азулен . [21] [22] Рефераты многих более ранних разработок в теории ab initio были опубликованы Шефером. [23]
В 1964 году были проведены расчеты методом Хюкеля (с использованием метода простой линейной комбинации атомных орбиталей (LCAO) для определения энергии электронов молекулярных орбиталей π-электронов в сопряженных углеводородных системах) молекул, сложность которых варьируется от бутадиена и бензола до овалена . компьютеры в Беркли и Оксфорде. [24] Эти эмпирические методы были заменены в 1960-х годах полуэмпирическими методами, такими как CNDO . [25]
В начале 1970-х годов для ускорения ab initio расчетов молекулярных орбиталей стали использоваться эффективные компьютерные программы ab initio , такие как ATMOL, Gaussian , IBMOL и POLYAYTOM. [26] Из этих четырех программ только Gaussian, которая в настоящее время значительно расширена, все еще используется, но сейчас используются и многие другие программы. [26] В то же время методы молекулярной механики , такие как силовое поле ММ2 , были разработаны, в первую очередь, Норманом Аллинджером . [27]
Одно из первых упоминаний термина « вычислительная химия» можно найти в книге Сидни Фернбаха и Абрахама Хаскелла Тауба «Компьютеры и их роль в физических науках» 1970 года, где они заявляют: «Поэтому кажется, что «вычислительная химия» наконец-то может быть все больше и больше становится реальностью». [28] В 1970-е годы самые разные методы стали рассматриваться как часть новой развивающейся дисциплины — вычислительной химии . [29] Журнал вычислительной химии был впервые опубликован в 1980 году.
Вычислительная химия была удостоена нескольких Нобелевских премий, особенно в 1998 и 2013 годах. Уолтер Кон «за разработку теории функционала плотности» и Джон Попл «за разработку вычислительных методов в квантовой химии» получили премию Нобелевская премия по химии 1998 года . [30] Мартин Карплюс , Майкл Левитт и Арье Варшел получили Нобелевскую премию по химии 2013 года за «разработку многомасштабных моделей сложных химических систем». [31]
В вычислительной химии есть несколько областей.
Эти поля могут привести к нескольким приложениям, как показано ниже.

Вычислительная химия — это инструмент для анализа каталитических систем без проведения экспериментов. Современная теория электронной структуры и теория функционала плотности позволили исследователям открыть и понять катализаторы . [37] Компьютерные исследования применяют теоретическую химию к исследованиям в области катализа. Методы теории функционала плотности рассчитывают энергии и орбитали молекул, чтобы создать модели этих структур. [38] Используя эти методы, исследователи могут прогнозировать такие значения, как энергия активации , реактивность сайта [39] и другие термодинамические свойства. [38]
Данные, которые сложно получить экспериментально, можно найти с помощью вычислительных методов моделирования механизмов каталитических циклов. [39] Квалифицированные компьютерные химики дают прогнозы, близкие к экспериментальным данным, при правильном учете методов и базисных наборов. Имея хорошие вычислительные данные, исследователи могут предсказать, как можно улучшить катализаторы, чтобы снизить стоимость и повысить эффективность этих реакций. [38]
Вычислительная химия используется при разработке лекарств для моделирования потенциально полезных молекул лекарств и помогает компаниям сэкономить время и деньги при разработке лекарств. Процесс открытия лекарств включает в себя анализ данных, поиск способов улучшения существующих молекул, поиск синтетических путей и тестирование этих молекул. [36] Вычислительная химия помогает в этом процессе, предсказывая, какие эксперименты лучше всего проводить без проведения других экспериментов. Вычислительные методы также позволяют найти значения, которые трудно найти экспериментально, например, pKa соединений. [40] Такие методы, как теория функционала плотности, можно использовать для моделирования молекул лекарств и определения их свойств, таких как энергии ВЗМО и НСМО, а также молекулярные орбитали. Компьютерные химики также помогают компаниям в разработке информатики, инфраструктуры и разработке лекарств. [41]
Помимо синтеза лекарств, носители лекарств также исследуются компьютерными химиками на предмет наноматериалов . Это позволяет исследователям моделировать окружающую среду для проверки эффективности и стабильности носителей лекарств. Понимание того, как вода взаимодействует с этими наноматериалами, обеспечивает стабильность материала в организме человека. Такое компьютерное моделирование помогает исследователям оптимизировать материал и найти лучший способ структурировать эти наноматериалы перед их созданием. [42]
Базы данных полезны как вычислительным, так и невычислительным химикам при исследованиях и проверке достоверности вычислительных методов. Эмпирические данные используются для анализа погрешности вычислительных методов по сравнению с экспериментальными данными. Эмпирические данные помогают исследователям с их методами и базисными наборами быть более уверенными в результатах исследователей. Базы данных вычислительной химии также используются при тестировании программного обеспечения или оборудования для вычислительной химии. [43]
Базы данных также могут использовать чисто расчетные данные. В чисто расчетных данных используются расчетные значения, а не экспериментальные значения для баз данных. Чисто рассчитанные данные позволяют избежать этих поправок на различные экспериментальные условия, такие как энергия нулевой точки. Эти расчеты также позволяют избежать экспериментальных ошибок для трудно поддающихся тестированию молекул. Хотя чисто расчетные данные часто не идеальны, выявлять проблемы часто легче на расчетных данных, чем на экспериментальных. [43]
Базы данных также предоставляют публичный доступ к информации, которую могут использовать исследователи. Они содержат данные, которые другие исследователи нашли и загрузили в эти базы данных, чтобы каждый мог их найти. Исследователи используют эти базы данных, чтобы найти информацию об интересующих молекулах и узнать, что можно сделать с этими молекулами. Некоторые общедоступные химические базы данных включают следующее. [43]
Программы, используемые в вычислительной химии, основаны на множестве различных квантово-химических методов, которые решают молекулярное уравнение Шрёдингера , связанное с молекулярным гамильтонианом . [46] Методы, которые не включают в свои уравнения какие-либо эмпирические или полуэмпирические параметры – будучи выведены непосредственно из теории, без включения экспериментальных данных – называются методами ab initio . [47] Теоретическое приближение строго определяется на основе первых принципов, а затем решается в пределах погрешности, которая качественно известна заранее. Если необходимо использовать числовые итерационные методы, цель состоит в том, чтобы выполнять итерации до тех пор, пока не будет получена полная машинная точность (наилучшее, что возможно при конечной длине слова на компьютере и в пределах сделанных математических и/или физических приближений). [48]
Методы ab initio должны определять уровень теории (метод) и базисный набор. [49] Базисный набор состоит из функций, сосредоточенных на атомах молекулы. Эти наборы затем используются для описания молекулярных орбиталей с помощью метода молекулярных орбиталей линейной комбинации атомных орбиталей (LCAO) ansatz . [50]

Распространенным типом расчета электронной структуры ab initio является метод Хартри-Фока (HF), расширение теории молекулярных орбиталей , где электрон-электронные отталкивания в молекуле специально не учитываются; в расчет учитывается только средний эффект электронов. По мере увеличения размера базисного набора энергия и волновая функция стремятся к пределу, называемому пределом Хартри – Фока. [50]
Многие типы расчетов начинаются с расчета Хартри-Фока и впоследствии корректируются с учетом электрон-электронного отталкивания, называемого также электронной корреляцией . [51] Эти типы вычислений называются пост-Хартри-Фоковскими методами. Постоянно совершенствуя эти методы, ученые могут все больше приближаться к идеальному предсказанию поведения атомных и молекулярных систем в рамках квантовой механики, как это определено уравнением Шредингера. [52] Чтобы получить точное согласие с экспериментом, необходимо включить специфические члены, некоторые из которых гораздо более важны для тяжелых атомов, чем для легких. [53]
В большинстве случаев волновая функция Хартри – Фока занимает одну конфигурацию или определитель. [54] В некоторых случаях, особенно для процессов разрыва связей, этого недостаточно, и необходимо использовать несколько конфигураций . [55]
Полную молекулярную энергию можно оценить как функцию молекулярной геометрии ; другими словами, поверхность потенциальной энергии . [56] Такую поверхность можно использовать для динамики реакций. Стационарные точки поверхности позволяют предсказать различные изомеры и переходные структуры для превращения между изомерами, но их можно определить без полного знания всей поверхности. [53]

Особенно важной задачей, называемой вычислительной термохимией , является расчет термохимических величин, таких как энтальпия образования, с химической точностью. Химическая точность — это точность, необходимая для реалистичных химических прогнозов, и обычно она составляет 1 ккал/моль или 4 кДж/моль. Чтобы достичь такой точности экономичным способом, необходимо использовать ряд пост-Хартри-Фоковских методов и объединить результаты. Эти методы называются композитными методами квантовой химии . [57]
После разделения электронных и ядерных переменных в рамках представления Борна – Оппенгеймера волновой пакет , соответствующий ядерным степеням свободы , распространяется через оператор временной эволюции (физика), связанный с нестационарным уравнением Шредингера (для полного молекулярного гамильтониана ). [58] В дополнительном энергозависимом подходе независимое от времени уравнение Шредингера решается с использованием формализма теории рассеяния . Потенциал, представляющий межатомное взаимодействие, задается поверхностями потенциальной энергии . В общем, поверхности потенциальной энергии связаны посредством условий вибронной связи . [59]
Наиболее популярными методами распространения волнового пакета , связанными с молекулярной геометрией, являются:
То, как вычислительный метод решает квантовые уравнения, влияет на точность и эффективность метода. Техника оператора расщепления является одним из таких методов решения дифференциальных уравнений. В вычислительной химии метод разделения операторов снижает вычислительные затраты на моделирование химических систем. Вычислительные затраты связаны с тем, сколько времени требуется компьютерам для расчета этих химических систем, тогда как для более сложных систем это может занять несколько дней. Квантовые системы сложны и требуют много времени для решения человеком. Методы оператора разделения помогают компьютерам быстро рассчитывать эти системы, решая подзадачи в квантовом дифференциальном уравнении . Метод делает это путем разделения дифференциального уравнения на два разных уравнения, например, когда имеется более двух операторов. После решения разделенные уравнения снова объединяются в одно уравнение, чтобы получить легко вычислимое решение. [62]
Этот метод используется во многих областях, требующих решения дифференциальных уравнений, например в биологии . Однако этот метод имеет ошибку разделения. Например, со следующим решением дифференциального уравнения. [62]
Уравнение можно разделить, но решения не будут точными, а только похожими. Это пример расщепления первого порядка. [62]
Есть способы уменьшить эту ошибку, в том числе взять среднее значение двух уравнений разделения. [62]
Другой способ повысить точность — использовать расщепление более высокого порядка. Обычно расщепление второго порядка — это самое большее, что делается, поскольку расщепление более высокого порядка требует гораздо больше времени для расчета и не стоит затрат. Методы более высокого порядка становятся слишком сложными для реализации и бесполезными для решения дифференциальных уравнений, несмотря на более высокую точность. [62]
Химики-вычислители тратят много времени на создание более точных систем, рассчитанных с помощью оператора разделения, при минимизации вычислительных затрат. Методы расчета являются серьезной проблемой для многих химиков, пытающихся моделировать молекулы или химическую среду. [62]

Методы теории функционала плотности (DFT) часто считаются методами ab initio для определения молекулярной электронной структуры, хотя многие из наиболее распространенных функционалов используют параметры, полученные на основе эмпирических данных или в результате более сложных расчетов. В ДПФ полная энергия выражается через полную одноэлектронную плотность, а не через волновую функцию. В этом типе расчета имеется приближенный гамильтониан и приближенное выражение для полной электронной плотности. Методы ДПФ могут быть очень точными при небольших вычислительных затратах. Некоторые методы сочетают в себе обменный функционал функционала плотности с обменным термином Хартри – Фока и называются гибридными функциональными методами. [63]
Полуэмпирические методы квантовой химии основаны на формализме метода Хартри-Фока , но используют множество приближений и получают некоторые параметры из эмпирических данных. Они были очень важны в вычислительной химии с 60-х по 90-е годы, особенно для лечения больших молекул, где полный метод Хартри-Фока без аппроксимаций был слишком дорогостоящим. Использование эмпирических параметров, по-видимому, позволяет в некоторой степени включить в методы корреляционные эффекты. [64]
Еще раньше были разработаны примитивные полуэмпирические методы, в которых двухэлектронная часть гамильтониана явно не учитывается. Для π-электронных систем это был метод Хюккеля, предложенный Эрихом Хюкелем , а для всех систем валентных электронов — расширенный метод Хюккеля , предложенный Роальдом Хоффманном . Иногда методы Хюккеля называют «полностью эмпирическими», поскольку они не выводятся из гамильтониана. [65] Тем не менее, термин «эмпирические методы» или «эмпирические силовые поля» обычно используется для описания молекулярной механики. [66]
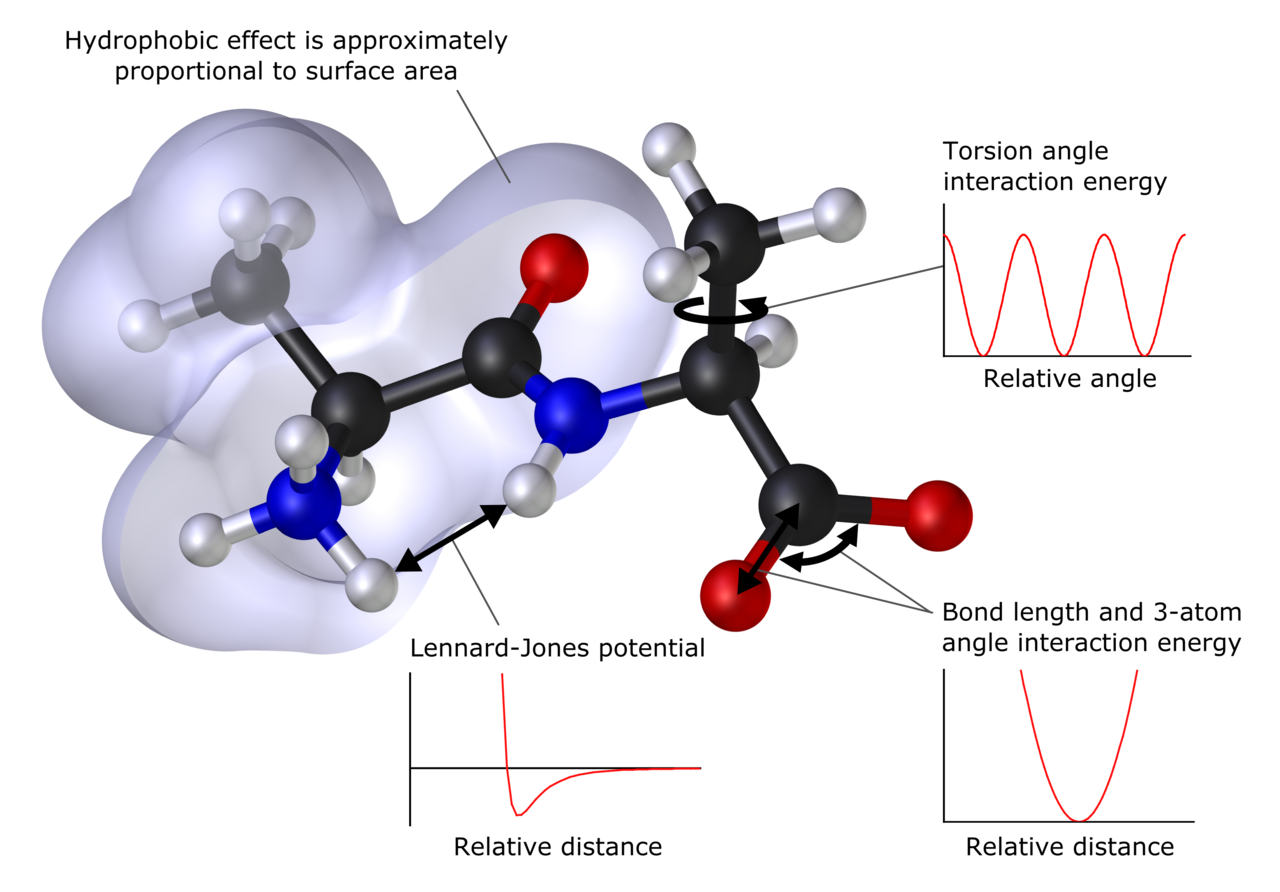
Во многих случаях большие молекулярные системы можно успешно моделировать, полностью избегая квантово-механических вычислений. Например, при моделировании молекулярной механики используется одно классическое выражение для энергии соединения, например, гармонический осциллятор . Все константы, входящие в уравнения, должны быть заранее получены из экспериментальных данных или расчетов ab initio . [64]
База данных соединений, используемых для параметризации, то есть результирующий набор параметров и функций, называемый силовым полем , имеет решающее значение для успеха расчетов молекулярной механики. Ожидается, что силовое поле, параметризованное против определенного класса молекул, например, белков, будет иметь какое-либо значение только при описании других молекул того же класса. [64] Эти методы могут быть применены к белкам и другим крупным биологическим молекулам и позволяют изучать сближение и взаимодействие (стыковку) потенциальных молекул лекарств. [67] [68]
Молекулярная динамика (МД) использует либо квантовую механику , либо молекулярную механику , либо их смесь для расчета сил, которые затем используются для решения законов движения Ньютона для изучения поведения систем, зависящего от времени. Результатом моделирования молекулярной динамики является траектория, описывающая, как положение и скорость частиц меняются со временем. Фазовая точка системы, описываемая положениями и импульсами всех ее частиц в предыдущий момент времени, определит следующую фазовую точку во времени путем интегрирования по законам движения Ньютона. [69]
Монте-Карло (MC) генерирует конфигурации системы, внося случайные изменения в положения ее частиц, а также их ориентацию и конформацию, где это необходимо. [70] Это метод случайной выборки, в котором используется так называемая выборка по важности . Методы выборки по важности способны генерировать состояния с низкой энергией, поскольку это позволяет точно рассчитывать свойства. Потенциальную энергию каждой конфигурации системы можно рассчитать вместе со значениями других свойств, исходя из положений атомов. [71] [72]
QM/MM — это гибридный метод, который пытается объединить точность квантовой механики со скоростью молекулярной механики. Это полезно для моделирования очень больших молекул, таких как ферменты . [73]
Квантовая вычислительная химия стремится использовать квантовые вычисления для моделирования химических систем, в отличие от подхода QM/MM (квантовая механика/молекулярная механика). [74] В то время как QM/MM использует гибридный подход, сочетающий квантовую механику для части системы с классической механикой для остальной части, квантовая вычислительная химия использует исключительно методы квантовых вычислений для представления и обработки информации, такие как гамильтоновы операторы. [75]
Обычные методы вычислительной химии часто сталкиваются со сложными квантово-механическими уравнениями, особенно из-за экспоненциального роста волновой функции квантовой системы. Квантовая вычислительная химия решает эти проблемы с помощью методов квантовых вычислений , таких как кубитизация и оценка квантовой фазы , которые, как полагают, предлагают масштабируемые решения. [76]
Кубитизация предполагает адаптацию оператора Гамильтона для более эффективной обработки на квантовых компьютерах, что повышает эффективность моделирования. С другой стороны, оценка квантовой фазы помогает точно определить собственные состояния энергии, которые имеют решающее значение для понимания поведения квантовой системы. [77]
Хотя эти методы продвинули область вычислительной химии, особенно в моделировании химических систем, их практическое применение в настоящее время ограничено в основном меньшими системами из-за технологических ограничений. Тем не менее, эти разработки могут привести к значительному прогрессу в достижении более точного и ресурсоэффективного моделирования квантовой химии. [76]
Вычислительные затраты и сложность алгоритмов в химии используются, чтобы помочь понять и предсказать химические явления. Они помогают определить, какие алгоритмы/вычислительные методы следует использовать при решении химических задач. В этом разделе основное внимание уделяется масштабированию вычислительной сложности в зависимости от размера молекул и подробному описанию алгоритмов, обычно используемых в обеих областях. [78]
В частности, в квантовой химии сложность может расти экспоненциально с увеличением количества электронов, участвующих в системе. Этот экспоненциальный рост является серьезным препятствием для точного моделирования больших или сложных систем. [79]
Передовые алгоритмы в обеих областях стремятся сбалансировать точность и эффективность вычислений. Например, в MD такие методы, как интеграция Верле или алгоритм Бимана, используются из-за их вычислительной эффективности. В квантовой химии гибридные методы, сочетающие различные вычислительные подходы (например, КМ/ММ), все чаще используются для решения больших биомолекулярных систем. [80]
Следующий список иллюстрирует влияние вычислительной сложности на алгоритмы, используемые в химических вычислениях. Важно отметить, что, хотя в этом списке представлены ключевые примеры, он не является исчерпывающим и служит руководством к пониманию того, как вычислительные требования влияют на выбор конкретных вычислительных методов в химии.
Решает уравнения движения Ньютона для атомов и молекул. [81]
Стандартный расчет парного взаимодействия в МД приводит к усложнению частиц. Это происходит потому, что каждая частица взаимодействует с каждой другой частицей, что приводит к взаимодействиям. [82] Усовершенствованные алгоритмы, такие как суммирование Эвальда или метод быстрых мультиполей, сводят это к группировке удаленных частиц или даже путем их обработки как единого целого или использования умных математических аппроксимаций. [83] [84]
Сочетает квантово-механические расчеты для небольшой области с молекулярной механикой для более крупной среды. [85]
Сложность методов КМ/ММ зависит как от размера квантовой области, так и от метода, используемого для квантовых расчетов. Например, если для квантовой части используется метод Хартри-Фока, сложность можно аппроксимировать как , где – количество базисных функций в квантовой области. Эта сложность возникает из-за необходимости итеративного решения набора связанных уравнений до тех пор, пока не будет достигнута самосогласованность. [86]

Находит одно состояние Фока, которое минимизирует энергию. [87]
NP-трудный или NP-полный, что продемонстрировано путем внедрения экземпляров модели Изинга в расчеты Хартри-Фока. Метод Хартри-Фока включает в себя решение уравнений Рутана-Холла, которые масштабируются в зависимости от реализации и количества базисных функций. Вычислительные затраты в основном связаны с оценкой и преобразованием двухэлектронных интегралов. Это доказательство NP-трудности или NP-полноты основано на внедрении таких проблем, как модель Изинга, в формализм Хартри-Фока. [87]
Исследует электронную структуру или ядерную структуру систем многих тел, таких как атомы, молекулы и конденсированные фазы . [89]
Традиционные реализации ДПФ обычно масштабируются как , в основном из-за необходимости диагонализации матрицы Кона-Шэма . [90] Шаг диагонализации, который находит собственные значения и собственные векторы матрицы, вносит наибольший вклад в это масштабирование. Недавние достижения в области ДПФ направлены на уменьшение этой сложности за счет различных аппроксимаций и усовершенствований алгоритмов. [91]
Методы CCSD и CCSD(T) представляют собой усовершенствованные методы электронной структуры, включающие одиночные, двойные, а в случае CCSD(T) пертурбативные тройные возбуждения для расчета эффектов электронной корреляции. [92]
Масштабируется по принципу: где – количество базисных функций. Эта интенсивная вычислительная потребность возникает из-за включения одиночных и двойных возбуждений в расчет электронной корреляции. [92]
С добавлением пертурбативных троек сложность возрастает до . Эта повышенная сложность ограничивает практическое использование меньшими системами, обычно до 20-25 атомов в традиционных реализациях. [92]

Адаптация стандартного метода CCSD(T) с использованием локальных естественных орбиталей (NO) для значительного снижения вычислительной нагрузки и возможности применения в более крупных системах. [92]
Обеспечивает линейное масштабирование в зависимости от размера системы, что является значительным улучшением по сравнению с традиционным масштабированием в пятой степени CCSD. Это достижение позволяет применять практические приложения к молекулам, насчитывающим до 100 атомов, с разумным базисным набором, что знаменует собой значительный шаг вперед в возможностях вычислительной химии обрабатывать более крупные системы с высокой точностью. [92]
Доказательство классов сложности алгоритмов включает в себя сочетание математических доказательств и вычислительных экспериментов. Например, в случае метода Хартри-Фока доказательство NP-трудности представляет собой теоретический результат, полученный из теории сложности, в частности, путем редукции известных NP-трудных задач. [93]
Для других методов, таких как MD или DFT, сложность вычислений часто наблюдается эмпирически и подтверждается анализом алгоритмов. В этих случаях доказательство правильности заключается не столько в формальных математических доказательствах, сколько в последовательном наблюдении за поведением вычислений в различных системах и реализациях. [93]
Вычислительная химия не является точным описанием реальной химии, поскольку математические и физические модели природы могут дать лишь приближение. Однако большинство химических явлений можно в той или иной степени описать качественной или приближенно-количественной вычислительной схемой. [94]
Молекулы состоят из ядер и электронов, поэтому применяются методы квантовой механики . Вычислительные химики часто пытаются решить нерелятивистское уравнение Шредингера с добавлением релятивистских поправок, хотя некоторый прогресс был достигнут в решении полностью релятивистского уравнения Дирака . В принципе, можно решить уравнение Шредингера либо в его нестационарной, либо нестационарной форме, в зависимости от решаемой задачи; на практике это невозможно, за исключением очень маленьких систем. Поэтому большое количество приближенных методов стремятся достичь наилучшего компромисса между точностью и вычислительными затратами. [95]
Точность всегда можно повысить за счет увеличения вычислительных затрат. Значительные ошибки могут возникнуть в ab initio моделях, содержащих много электронов, из-за вычислительной стоимости полностью релятивистских методов. [92] Это усложняет изучение молекул, взаимодействующих с атомами большой атомной массы, таких как переходные металлы, и их каталитических свойств. Современные алгоритмы вычислительной химии позволяют регулярно рассчитывать свойства небольших молекул, содержащих до 40 электронов, с ошибками для энергий менее нескольких кДж/моль. Для геометрических фигур длины связей можно предсказать в пределах нескольких пикометров, а валентные углы - в пределах 0,5 градуса. Обработку более крупных молекул, содержащих несколько десятков атомов, можно выполнить с помощью вычислений с помощью более приближенных методов, таких как теория функционала плотности (DFT). [96]
В этой области ведутся споры о том, достаточны ли последние методы для описания сложных химических реакций, например, в биохимии. Большие молекулы можно изучать полуэмпирическими приближенными методами. Даже более крупные молекулы обрабатываются методами классической механики , которые используют так называемую молекулярную механику (ММ). В методах КМ-ММ небольшие части больших комплексов обрабатываются квантово-механически (КМ), а остальная часть обрабатывается приближенно (ММ). [97]
Существует множество самодостаточных пакетов программного обеспечения для вычислительной химии . Некоторые включают множество методов, охватывающих широкий диапазон, тогда как другие концентрируются на очень конкретном диапазоне или даже на одном методе. Подробности о большинстве из них можно найти в:











