Борон, Уолтер Ф.; Булпаеп, Эмиль Л., ред. (2017). Медицинская физиология (3-е изд.). Филадельфия, Пенсильвания: Elsevier. ISBN 978-1-4557-4377-3.

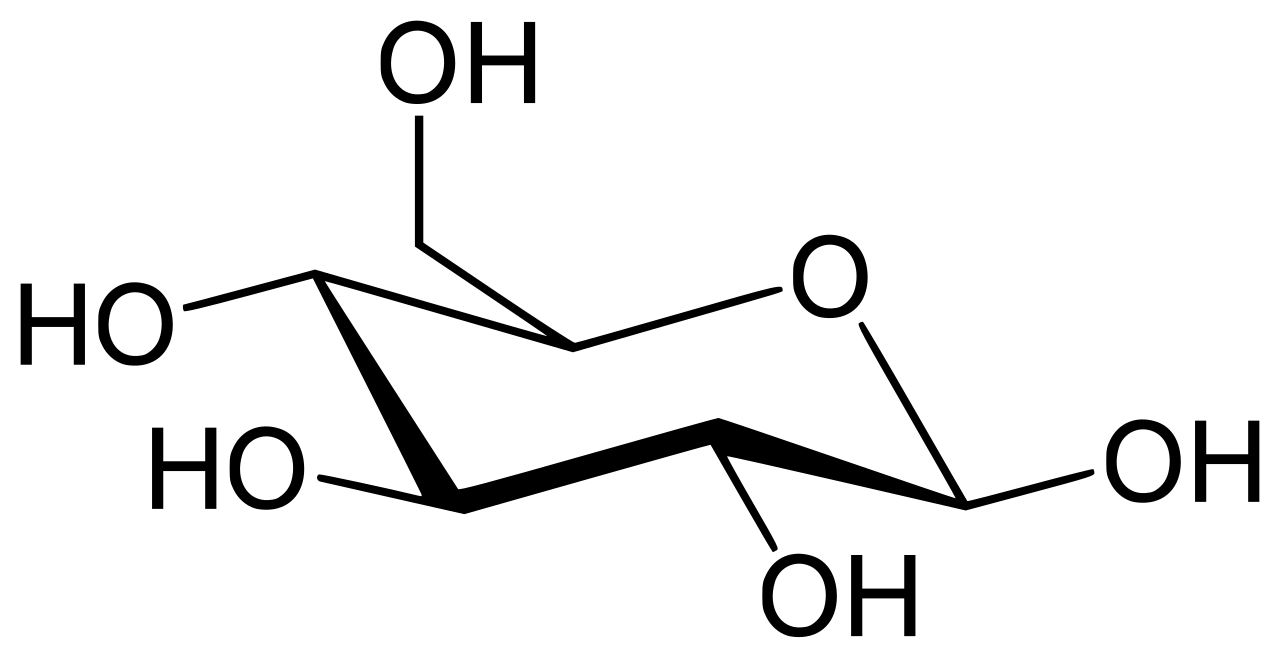
Фермент глюкозо-6-фосфатаза (EC 3.1.3.9, G6Pase ; систематическое название D -глюкозо-6-фосфатфосфогидролаза ) катализирует гидролиз глюкозо-6-фосфата , что приводит к образованию фосфатной группы и свободной глюкозы:
Во время голодания адекватный уровень глюкозы в крови обеспечивается глюкозой, высвобождаемой из запасов гликогена печени путем гликогенолиза , а также глюкозой, вырабатываемой глюконеогенезом в печени, а также — в меньшей степени — в почках. G6P является продуктом обоих этих путей [1] и должен быть преобразован в глюкозу, прежде чем он сможет быть экспортирован из клетки в кровь мембраносвязанными переносчиками глюкозы . [2] Таким образом, G6Pase в основном экспрессируется в печени и почках [1] — в то время как скелетные мышцы в совокупности содержат наиболее существенный запас гликогена в организме, глюкоза не может быть мобилизована из них, поскольку в мышцах отсутствует G6Pase. [3] : 1171
Инсулин подавляет активность печеночной G6Pase, [3] : 1046, тогда как глюкагон стимулирует ее. [3] : 1052 Экспрессия G6Pase увеличивается при голодании, при диабете и при приеме глюкокортикостероидов . [1]
Глюкозо-6-фосфатаза представляет собой комплекс многокомпонентных белков, включая транспортеры для G6P, глюкозы и фосфата. Основная функция фосфатазы выполняется каталитической субъединицей глюкозо-6-фосфатазы. У человека существует три изофермента каталитической субъединицы: глюкозо-6-фосфатаза-α, кодируемая G6PC ; IGRP, кодируемая G6PC2 ; и глюкозо-6-фосфатаза-β, кодируемая G6PC3 . [4]
Глюкозо-6-фосфатаза-α и глюкозо-6-фосфатаза-β являются функциональными фосфогидролазами и имеют схожую структуру активного центра, топологию, механизм действия и кинетические свойства в отношении гидролиза G6P. [5] Напротив, IGRP практически не имеет гидролазной активности и может играть другую роль в стимуляции секреции инсулина поджелудочной железой. [6]
.png/1280px-Wiki_image_(2).png)
.png/1280px-Final_for_wiki_(1).png)

Хотя четкий консенсус не был достигнут, большое количество ученых придерживаются модели транспорта субстрата для учета каталитических свойств глюкозо-6-фосфатазы. В этой модели глюкозо-6-фосфатаза имеет низкую степень селективности. Перенос глюкозо-6-фосфата осуществляется транспортным белком (Т1), а эндоплазматический ретикулум (ЭР) содержит структуры, позволяющие выход фосфатной группы (Т2) и глюкозы (Т3). [7]
Глюкозо-6-фосфатаза состоит из 357 аминокислот и прикреплена к эндоплазматическому ретикулуму (ЭР) девятью трансмембранными спиралями. Ее N -конец и активный центр находятся на стороне просвета ЭР, а ее С -конец выступает в цитоплазму. Из-за ее тесной связи с ЭР точная структура глюкозо-6-фосфатазы остается неизвестной. Однако выравнивание последовательностей показало, что глюкозо-6-фосфатаза структурно похожа на активный центр хлорпероксидазы, содержащей ванадий, обнаруженной в Curvularia inaequalis. [8]
На основе исследований кинетики pH катализа глюкозо-6-фосфатазы-α было высказано предположение, что гидролиз глюкозо-6-фосфата завершается через ковалентный фосфогистидин глюкозо-6-фосфатный интермедиат. Активный сайт глюкозо-6-фосфатазы-α был первоначально идентифицирован по наличию консервативного мотива фосфатной сигнатуры, обычно обнаруживаемого в липидных фосфатазах, кислых фосфатазах и ванадиевых галопероксидазах. [5]
Основные остатки в активном центре ванадиевых галопероксидаз включают: Lys353, Arg360, Arg490, His404 и His496. Соответствующие остатки в активном центре глюкозо-6-фосфатазы-α включают Arg170 и Arg83, которые отдают ионы водорода фосфату, стабилизируя переходное состояние, His119, который обеспечивает протон для дефосфорилированного кислорода, присоединенного к глюкозе, и His176, который завершает нуклеофильную атаку на фосфат с образованием ковалентно связанного фосфорилированного промежуточного фермента. [9] В хлоропероксидазе, содержащей ванадий, было обнаружено, что Lys353 стабилизирует фосфат в переходном состоянии. Однако соответствующий остаток в глюкозо-6-фосфатазе-α (Lys76) находится внутри мембраны ЭР, и его функция, если таковая имеется, в настоящее время не определена. За исключением Lys76, все эти остатки расположены на люминальной стороне мембраны ЭР. [5]
Глюкозо-6-фосфатаза-β — это повсеместно экспрессируемый мембранный белок из 346 аминокислот, который на 36% идентичен последовательности глюкозо-6-фосфатазы-α. В ферменте глюкозо-6-фосфатазы-β выравнивание последовательностей предсказывает, что его активный центр содержит His167, His114 и Arg79. Подобно активному центру глюкозо-6-фосфатазы-α, His167 — это остаток, который обеспечивает нуклеофильную атаку, а His114 и Arg79 — доноры водорода. Глюкозо-6-фосфатаза-β также локализована в мембране ЭР, хотя ее ориентация неизвестна. [5]
Гидролиз глюкозо-6-фосфата начинается с нуклеофильной атаки His176 на связанный с сахаром фосфат, что приводит к образованию фосфогистидиновой связи и деградации карбонила. Затем отрицательно заряженный кислород передает свои электроны, преобразуя карбонил и разрывая его связь с глюкозой. Затем отрицательно заряженный связанный с глюкозой кислород протонируется His119, образуя свободную глюкозу. Фосфо-промежуточное соединение, полученное в результате реакции между His176 и фосфатной группой, затем разрушается гидрофильной атакой; после добавления другого гидроксида и разложения карбонила карбонил преобразуется, отнимая электроны, первоначально предоставленные остатком His176, тем самым создавая свободную фосфатную группу и завершая гидролиз. [9]
Гены, кодирующие фермент, в первую очередь экспрессируются в печени, корковом веществе почек и (в меньшей степени) в β-клетках островков поджелудочной железы и слизистой оболочке кишечника (особенно во время голодания). [7] Глюкозо-6-фосфатаза присутствует в самых разных мышцах животного мира, хотя и в очень низких концентрациях. [10] Таким образом, гликоген, который запасают мышцы, обычно недоступен для остальных клеток организма, поскольку глюкозо-6-фосфат не может пересечь сарколемму, если он не дефосфорилирован. Фермент играет важную роль в периоды голодания и при низком уровне глюкозы. Было показано, что голодание и диабет вызывают двух-трехкратное увеличение активности глюкозо-6-фосфатазы в печени. [7] Активность Glc 6-Pase также резко возрастает при рождении, когда организм становится независимым от источника глюкозы матери. Ген человеческого Glc 6-Pase содержит пять экзонов, охватывающих приблизительно 125,5 кб ДНК, расположенных на хромосоме 17q21. [11]
Мутации системы глюкозо-6-фосфатазы, а именно субъединицы глюкозо-6-фосфатазы-α (глюкозо-6-фосфатаза-α), транспортера глюкозы-6 (G6PT) и субъединицы глюкозо-6-фосфатазы-β (глюкозо-6-фосфатаза-β или G6PC3), приводят к дефициту поддержания интерпрандиального гомеостаза глюкозы , а также функции и гомеостаза нейтрофилов . [12] [13] Мутации как в глюкозо-6-фосфатазе-α, так и в G6PT приводят к болезни накопления гликогена I типа (GSD 1, болезнь фон Гирке). [14] А именно, мутации в глюкозо-6-фосфатазе-α приводят к болезни накопления гликогена I типа, которая характеризуется накоплением гликогена и жира в печени и почках, что приводит к гепатомегалии и реномегалии. [15] GSD-1a составляет приблизительно 80% случаев GSD-1, которые проявляются клинически. [16] Отсутствие G6PT приводит к GSD-1b (GSD-1b), который характеризуется отсутствием G6PT и составляет 20% случаев, которые проявляются клинически. [16] [17]

Конкретная причина GSD-1a вытекает из бессмысленных мутаций, вставок/делеций со сдвигом рамки считывания или без него или мутаций сайта сплайсинга, которые происходят на генетическом уровне. [7] Миссенс-мутации затрагивают две большие просветные петли и трансмембранные спирали глюкозо-6-фосфатазы-α, отменяя или значительно снижая активность фермента. [7] Конкретная причина GSD-1b вытекает из «тяжелых» мутаций, таких как мутации сайта сплайсинга, мутации со сдвигом рамки и замены высококонсервативного остатка, которые полностью разрушают активность G6PT. [7] Эти мутации приводят к распространению GSD-1, предотвращая транспорт глюкозо-6-фосфата (G6P) в просветную часть ЭР , а также ингибируя превращение G6P в глюкозу для использования клеткой.
Третий тип дефицита глюкозо-6-фосфатазы, дефицит глюкозо-6-фосфатазы-β, характеризуется синдромом врожденной нейтропении , при котором нейтрофилы демонстрируют повышенный стресс эндоплазматического ретикулума (ЭР), повышенный апоптоз, нарушенный энергетический гомеостаз и нарушенную функциональность. [18] Он также может привести к сердечным и урогенитальным порокам развития. [19] Этот третий класс дефицита также зависит от дефицита G6PT, поскольку глюкозо-6-фосфатаза-β также находится в просвете ЭР и, таким образом, может приводить к схожим симптомам дефицита глюкозо-6-фосфатазы-β, связанным с GSD-1b. [17] Кроме того, недавние исследования выявили эту область сходства между обоими дефицитами и показали, что аберрантное гликозилирование происходит при обоих дефицитах. [20] Гликозилирование нейтрофилов оказывает сильное влияние на активность нейтрофилов и, таким образом, может также классифицироваться как врожденное нарушение гликозилирования. [20]
Основная функция глюкозо-6-фосфатазы-β была определена как обеспечение переработанной глюкозы в цитоплазме нейтрофилов для поддержания нормальной функции. Нарушение соотношения глюкозы к G6P из-за значительного снижения внутриклеточных уровней глюкозы вызывает значительное нарушение гликолиза и HMS . [ 13] Если не противодействовать поглощению внеклеточной глюкозы, этот дефицит приводит к дисфункции нейтрофилов. [13]
Было показано, что соединения ванадия, такие как сульфат ванадила, ингибируют фермент и, таким образом, повышают чувствительность к инсулину in vivo у диабетиков, что было оценено с помощью метода гиперинсулинемического зажима , что может иметь потенциальные терапевтические последствия. [21] [22]
Молекулярные графические изображения были получены с использованием UCSF Chimera. [23]
{{cite journal}}: Цитировать журнал требует |journal=( помощь ){kind=link}