Талассемия — это наследственное заболевание крови , которое приводит к аномальному гемоглобину . [7] Симптомы зависят от типа талассемии и могут варьироваться от отсутствия до тяжелых. [1] Часто наблюдается анемия от легкой до тяжелой степени (низкий уровень эритроцитов или гемоглобина), поскольку талассемия может влиять на выработку эритроцитов, а также на продолжительность жизни эритроцитов. [1] Симптомы анемии включают чувство усталости и бледность кожи . [1] Другие симптомы талассемии включают проблемы с костями, увеличение селезенки , желтоватую кожу , легочную гипертензию и темную мочу. [1] У детей может наблюдаться замедление роста. [1] Симптомы и проявления талассемии могут со временем меняться.
Лечение зависит от типа и степени тяжести. [4] Лечение людей с более тяжелым заболеванием часто включает регулярное переливание крови , хелатирование железа и фолиевую кислоту . [4] Хелатирование железа можно проводить с помощью дефероксамина , деферазирокса или деферипрона . [4] [9] Иногда вариантом может быть трансплантация костного мозга . [4] Осложнения могут включать перегрузку железом в результате переливания крови, приводящую к заболеваниям сердца или печени , инфекциям и остеопорозу . [1] Если селезенка чрезмерно увеличивается, может потребоваться хирургическое удаление . [1] Пациенты с талассемией, которые плохо реагируют на переливание крови, могут принимать гидроксимочевину или талидомид , а иногда и их комбинацию. [10] Гидроксимочевина — единственный препарат для лечения талассемии, одобренный FDA. Пациенты, принимавшие гидроксимочевину в дозе 10 мг/кг каждый день в течение года, имели значительно более высокий уровень гемоглобина, и это было хорошо переносимое лечение для пациентов, которые плохо реагировали на переливание крови. [11] Другие известные индукторы гемоглобина включают талидомид, но он не тестировался в клинических условиях. Комбинация талидомида и гидроксимочевины приводила к значительному повышению уровня гемоглобина у трансфузионно-зависимых и нетрансфузионно-зависимых пациентов [12]
По состоянию на 2015 год талассемия встречается примерно у 280 миллионов человек, из которых около 439 000 страдают тяжелым заболеванием. [13] Это наиболее распространено среди людей греческого , итальянского , ближневосточного , южноазиатского и африканского происхождения. [7] Мужчины и женщины имеют одинаковые показатели заболеваемости. [14] Это привело к 16 800 смертям в 2015 году по сравнению с 36 000 смертей в 1990 году. [6] [15] Те, у кого талассемия легкой степени, как и те, у кого есть серповидноклеточный признак , имеют некоторую защиту от малярии , что объясняет почему серповидноклеточная анемия и талассемия чаще встречаются в регионах мира, где риск малярии выше. [16] По оценкам, 1/3 людей с талассемией страдают «независимой от переливания крови талассемией», и их выживание не зависит от регулярного продолжения переливания крови.
Признаки и симптомы
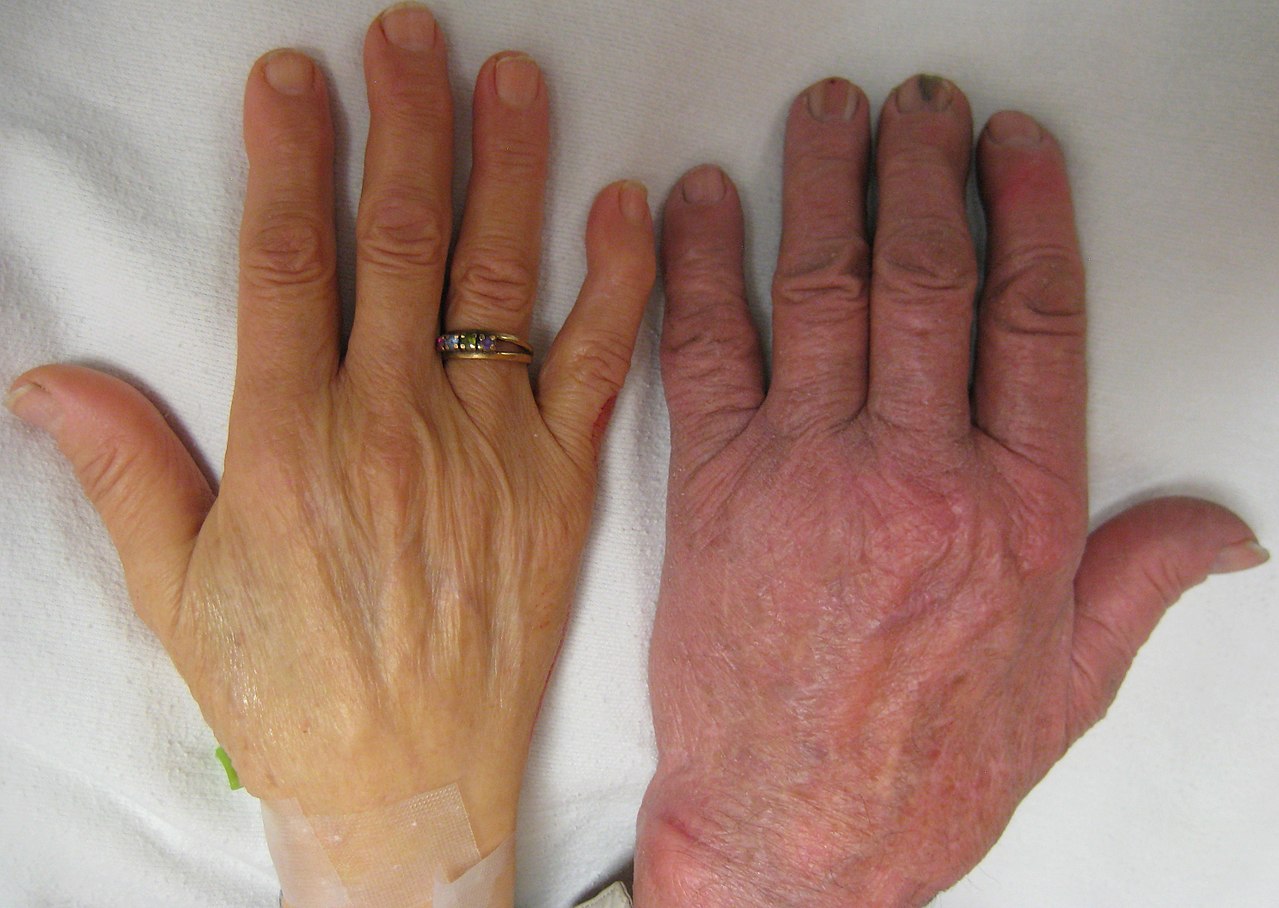
Слева: рука человека с тяжелой анемией. Справа: рука человека без анемии.У пациента с талассемией увеличена селезенка.
Перегрузка железом . Люди с талассемией могут получить перегрузку железа в организме либо из-за самой болезни, либо из-за частых переливаний крови. Слишком много железа может привести к повреждению сердца, печени и эндокринной системы , в том числе желез, вырабатывающих гормоны, регулирующие процессы во всем организме. Повреждение характеризуется чрезмерными отложениями железа. Без адекватной терапии хелатированием железа почти у всех пациентов с бета-талассемией накапливаются потенциально смертельные уровни железа. [17]
Инфекция. Люди с талассемией имеют повышенный риск заражения. Это особенно актуально, если селезенка была удалена. [18]
Деформации костей. Талассемия может привести к расширению костного мозга, что приводит к расширению костей. Это может привести к нарушению структуры костей, особенно лица и черепа. Расширение костного мозга также делает кости тонкими и ломкими, что увеличивает риск переломов костей. [19]
Увеличенная селезенка . Селезенка помогает бороться с инфекцией и фильтрует нежелательный материал, например, старые или поврежденные клетки крови. Талассемия часто сопровождается разрушением большого количества эритроцитов, и задача удаления этих клеток приводит к увеличению селезенки. Спленомегалия может усугубить анемию и сократить жизнь перелитых эритроцитов. Сильное увеличение селезенки может потребовать ее удаления. [20]
Замедление темпов роста: анемия может привести к замедлению роста ребенка. Половое созревание также может быть задержано у детей с талассемией. [21]
Проблемы с сердцем: такие заболевания, как застойная сердечная недостаточность и нарушения сердечного ритма, могут быть связаны с тяжелой талассемией. [22]
Структурная биология гемоглобина
Нормальные гемоглобины человека представляют собой тетрамерные белки, состоящие из двух пар цепей глобина, каждая из которых содержит одну альфа-подобную (α-подобную) цепь и одну бета-подобную (β-подобную) цепь. Каждая цепь глобина связана с железосодержащим фрагментом гема. На протяжении жизни синтез альфа-подобных и бета-подобных (также называемых неальфа-подобными) цепей сбалансирован, так что их соотношение относительно постоянно и не происходит избытка того или иного типа. [23]
Специфические альфа- и бета-подобные цепи, входящие в состав гемоглобина, строго регулируются во время развития:
Эмбриональные гемоглобины экспрессируются уже на 4-6 неделе эмбриогенеза и исчезают примерно на восьмой неделе беременности, когда они заменяются фетальными гемоглобинами. [24] [25] Эмбриональные Hbs включают:
Hb Gower-1, состоящий из двух ζ-глобинов (дзета-глобинов) и двух ε-глобинов (эпсилон-глобинов) (ζ2ε2)
Hb Gower-2, состоящий из двух альфа-глобинов и двух эпсилон-глобинов (α2ε2).
Hb Портленд, состоящий из двух зета-глобинов и двух гамма-глобинов (ζ2γ2).
Фетальный гемоглобин (Hb F) вырабатывается примерно с восьми недель беременности до рождения и составляет примерно 80 процентов гемоглобина у доношенного новорожденного. Он снижается в течение первых нескольких месяцев жизни и в нормальном состоянии составляет <1 процента от общего уровня гемоглобина к раннему детскому возрасту. Hb F состоит из двух альфа-глобинов и двух гамма-глобинов (α2γ2). У пациентов с β-талассемией наблюдаются более высокие уровни гамма-глобулина и, следовательно, большее производство Hb F, чтобы противодействовать дисбалансу, связанному с неспособностью производить бета-цепи. [26]
Уровень гемоглобина взрослых ( Hb A ) является преобладающим гемоглобином у детей в возрасте шести месяцев и старше; он составляет 96-97% от общего гемоглобина у лиц без гемоглобинопатии. Он состоит из двух альфа-глобинов и двух бета-глобинов (α2β2). [ нужна цитата ]
Hb A2 представляет собой второстепенный уровень Hb у взрослых, который обычно составляет примерно 2,5–3,5% от общего уровня Hb, начиная с шестимесячного возраста. Он состоит из двух альфа-глобинов и двух дельта-глобинов (α2δ2). [ нужна цитата ]
Как α-, так и β-талассемия часто наследуются по аутосомно- рецессивному типу. Сообщалось о случаях доминантно наследуемых α- и β-талассемий, первый из которых произошел в ирландской семье с двумя делециями 4 и 11 п.н. в экзоне 3, прерванными вставкой 5 п.н. в гене β-глобина. При аутосомно-рецессивных формах заболевания оба родителя должны быть носителями, чтобы ребенок мог заболеть. Если оба родителя являются носителями признаков гемоглобинопатии, риск составляет 25% для каждой беременности у больного ребенка. [27]
Гены, участвующие в талассемии, контролируют выработку здорового гемоглобина. Гемоглобин связывает кислород в легких и высвобождает его, когда эритроциты достигают периферических тканей, таких как печень. Связывание и высвобождение кислорода гемоглобином необходимы для выживания. [ нужна цитата ]
Эволюция
Наличие единственного генетического варианта талассемии может защитить от малярии и, таким образом, может быть преимуществом. [28]
В норме большая часть взрослого гемоглобина ( HbA ) состоит из четырех белковых цепей, двух α- и двух β-цепей глобина, образующих гетеротетрамер . При талассемии у пациентов наблюдаются дефекты либо в α-, либо в β-цепи глобина, вызывающие выработку аномальных эритроцитов. [ нужна цитата ]
Талассемии классифицируют в зависимости от того, какая цепь молекулы гемоглобина поражена. При α-талассемиях нарушается выработка цепи α-глобина, тогда как при β-талассемии нарушается выработка цепи β-глобина. [30]
Цепи β-глобина кодируются одним геном на хромосоме 11 ; Цепи α-глобина кодируются двумя тесно связанными генами на хромосоме 16 . [31] Таким образом, у нормального человека с двумя копиями каждой хромосомы два локуса кодируют β-цепь, а четыре локуса кодируют α-цепь. Делеция одного из α-локусов широко распространена у людей африканского или азиатского происхождения, что повышает вероятность развития α-талассемии. β-талассемия распространена не только у африканцев , но также у греков и турок . [ нужна цитата ]
Альфа-талассемия
В α-талассемии участвуют гены HBA1 [32] и HBA2 [33] , наследуемые по менделевскому рецессивному типу. Существует два генных локуса и, следовательно, четыре аллели. Для α-глобина существуют два генетических локуса, таким образом, в диплоидных клетках присутствуют четыре аллеля. Два аллеля имеют материнское происхождение, а два аллеля — отцовское. Тяжесть α-талассемий коррелирует с количеством пораженного α-глобина; аллели: чем больше, тем тяжелее будут проявления заболевания. [34] Альфа-талассемия приводит к снижению выработки альфа-глобина; следовательно, образуется меньше цепей альфа-глобина, что приводит к избытку β-цепей у взрослых и избытку γ-цепей у новорожденных. Избыточные β-цепи образуют нестабильные тетрамеры (называемые гемоглобином H или HbH из 4 бета-цепей), которые имеют аномальные кривые диссоциации кислорода. Альфа-талассемия часто встречается у людей из Юго-Восточной Азии, Ближнего Востока, Китая и у лиц африканского происхождения. [35]
Бета-талассемия
Бета-талассемия возникает из-за мутаций гена HBB на хромосоме 11 [36] , которые также наследуются по аутосомно-рецессивному типу. Тяжесть заболевания зависит от характера мутации и наличия мутаций в одном или обоих аллелях. Мутированные аллели называются β + , когда частичная функция сохраняется (либо белок имеет сниженную функцию, либо он функционирует нормально, но производится в уменьшенном количестве) или βo , когда функционирующий белок не вырабатывается. Положение обоих аллелей определяет клиническую картину:
Большая β-талассемия ( средиземноморская анемия или анемия Кули ) вызывается генотипом βo / βo . Функциональные β-цепи не образуются, и, следовательно, гемоглобин А не может быть собран. Это наиболее тяжелая форма β-талассемии;
Промежуточная β-талассемия обусловлена генотипом β + /β o или β + /β + . В этой форме вырабатывается некоторое количество гемоглобина А;
Малая β-талассемия обусловлена генотипом β/β o или β/β + . Только один из двух аллелей β-глобина содержит мутацию, поэтому выработка β-цепи не сильно нарушена, и пациенты могут протекать относительно бессимптомно.
Бета-талассемия чаще всего встречается у людей средиземноморского происхождения. В меньшей степени могут пострадать китайцы, другие азиаты и афроамериканцы. [35]
Дельта-талассемия
Помимо альфа- и бета-цепей, присутствующих в гемоглобине, около 3% гемоглобина взрослого человека состоит из альфа- и дельта-цепей. Как и в случае с бета-талассемией, могут возникать мутации, влияющие на способность этого гена производить дельта-цепи. [37] [38]
Комбинированные гемоглобинопатии
Талассемия может сосуществовать с другими гемоглобинопатиями . Наиболее распространенными из них являются:
Гемоглобин C /талассемия: распространен в средиземноморском и африканском населении, гемоглобин C/β талассемия вызывает умеренно тяжелую гемолитическую анемию со спленомегалией; гемоглобин C/β + талассемия вызывает более легкое заболевание. [ нужна цитата ]
Учитывая диапазон тяжести заболевания, некоторые люди не нуждаются в лечении (те, у кого нет симптомов), в то время как некоторым для выживания требуется регулярное переливание крови. [43] Людям с тяжелой формой талассемии требуется медицинское лечение, и основным методом лечения обычно является переливание эритроцитов.
Переливание эритроцитов
Переливание крови является основным методом лечения, позволяющим продлить жизнь. [44] Необходимый подход и частота варьируются у каждого человека в зависимости от тяжести заболевания, возраста, задержки роста, наличия экстрамедуллярного эритропоэза (педиатрия), беременности и состояния сердца. Переливание крови сопряжено с рисками, включая усугубление перегрузки железом, риск инфекций, риск образования антител к эритроцитам, повышенный риск развития реакций гиперчувствительности и риск воспаления желчного пузыря ( холецистита ). [43]
Многократные переливания крови могут привести к перегрузке железом. Перегрузку железом, связанную с талассемией, можно лечить с помощью хелаторной терапии с использованием препаратов дефероксамина , деферипрона или деферазирокса . [45] [46] [47] Эти методы лечения привели к увеличению продолжительности жизни людей с большой талассемией. [45] Дефероксамин эффективен только при ежедневной инъекции, что усложняет его длительное применение. Однако это недорого и безопасно. Побочные эффекты включают первичные кожные реакции вокруг места инъекции и потерю слуха . [45] Деферасирокс и деферипрон являются пероральными препаратами, общие побочные эффекты которых включают тошноту, рвоту и диарею. При сравнении эффективности нет доказательств превосходства деферазирокса или деферипрона, однако долгосрочное сравнение не проводилось. [47] Деферасирокс не эффективен для всех пациентов и может не подходить для тех, у кого серьезные проблемы с сердцем, связанные с перегрузкой железом, в то время как деферипрон, по-видимому, является наиболее эффективным средством при поражении сердца. Кроме того, стоимость деферазирокса также значительна. [45] Сочетание блокаторов кальциевых каналов с терапией хелатирования железа находится в стадии изучения, однако преимущества не ясны из проведенных клинических испытаний. [48]
Трансплантация костного мозга может дать возможность излечения молодых людей, имеющих HLA -совместимого донора. [50] Показатели успеха достигли диапазона 80–90%. [50] Смертность от процедуры составляет около 3%. [51] Рандомизированных контролируемых исследований, в которых проверялась бы безопасность и эффективность трансплантации костного мозга неидентичных доноров у лиц с β-талассемией, зависимых от переливания крови, не проводилось. [52]
Болезни «трансплантат против хозяина» (РТПХ) являются одним из важных побочных эффектов трансплантации костного мозга. Необходимы дальнейшие исследования, чтобы оценить, можно ли использовать мезенхимальные стромальные клетки в качестве профилактики или лечения РТПХ. [53]
Если у человека нет совместимого по HLA донора, можно попытаться провести трансплантацию костного мозга от гаплоидентичной матери ребенку (несовместимому донору). В исследовании с участием 31 человека выживаемость без талассемии составила 70%, отторжение — 23%, а смертность — 7%. Наиболее положительные результаты обычно наблюдаются у очень молодых людей. [54]
Другие методы лечения
Многие методы лечения исследуются с целью сокращения количества переливаний крови, необходимых человеку. Лекарства, индуцирующие фетальный гемоглобин (HbF), были опробованы с целью повышения уровня гемоглобина и уменьшения зависимости человека от переливания крови, однако польза неясна, и ранние результаты не показывают клинически значимого преимущества. [43] Лечение гидроксимочевиной с целью реактивации гамма-генов для производства HbF также не имеет доказательств высокого качества, подтверждающих его эффективность. [55]
В результате рандомизированных контролируемых исследований нет доказательств в поддержку приема добавок цинка у людей с талассемией. [56] Компьютерные программы или мобильные приложения были предложены в качестве инструментов, помогающих людям управлять талассемией и следовать лечению, включая терапию хелатированием железа. Эффективность этих приложений недостаточно исследована. [57]
Люди с талассемией подвергаются более высокому риску развития остеопороза. [58] Варианты лечения включают бисфосфонаты, а иногда и гормональную терапию. Были предложены другие методы лечения, включая кальцитонин, цинк, гидроксимочевину и добавки кальция. Эффективность бисфосфонатов и цинка не ясна, и необходимы дальнейшие исследования. [58]
Стоматологическая помощь
Помощь людям с талассемией в уходе за зубами и лечении стоматологических проблем может быть сложной задачей из-за основного заболевания. Перегрузка железом из-за переливания крови может привести к отложению железа в зубах и изменению их цвета, а также существует повышенный риск заражения. [59] Варианты лечения людей с талассемией необходимо изменить, чтобы обеспечить учет потребностей человека, а также настоятельно рекомендуется раннее выявление и раннее лечение любых проблем, чтобы снизить риск того, что человеку потребуется более сложное стоматологическое лечение. [59] Доказательства, подтверждающие эффективность стоматологического лечения у людей с талассемией, слабы, и необходимы более качественные клинические исследования. [59]
Легкая талассемия
Людям с признаками талассемии не требуется медицинская или последующая помощь после постановки первоначального диагноза. [44] Людей с признаком β-талассемии следует предупредить, что их состояние может быть ошибочно принято за более распространенную железодефицитную анемию . Им следует избегать регулярного приема добавок железа , но дефицит железа может развиться во время беременности или в результате хронического кровотечения. [60] Генетическое консультирование показано всем лицам с генетическими нарушениями, особенно если в семье существует риск тяжелой формы заболевания, которую можно предотвратить. [61]
Профилактика
Американский колледж акушеров и гинекологов рекомендует всем людям, планирующим беременность, пройти тестирование на наличие талассемии. [62] Генетическое консультирование и генетическое тестирование рекомендуются семьям, имеющим признаки талассемии. [27] Мы надеемся, что понимание генетического риска, в идеале до создания семьи, позволит семьям больше понять об этом заболевании и принять обоснованное решение, которое лучше всего подходит для их семьи. [27]
На Кипре существует политика скрининга , направленная на снижение заболеваемости талассемией, которая с момента реализации программы в 1970-х годах (включая пренатальный скрининг и аборты) снизила количество детей, рожденных с этим заболеванием, с одного из каждых 158 рождений почти до нуля. . [63] В Греции также существует программа скрининга для выявления людей, являющихся носителями. [64]
В Иране в качестве добрачного скрининга сначала проверяются показатели эритроцитов мужчины. Если у него имеется микроцитоз ( средний гемоглобин в клетке < 27 пг или средний объем эритроцитов < 80 мкл), женщину обследуют. Когда оба являются микроцитарными, измеряют концентрацию гемоглобина А2 . Если у обоих концентрация превышает 3,5% (диагностический признак талассемии), их направляют в местный назначенный медицинский пункт для генетической консультации . [65]
В Индии как правительственными, так и неправительственными организациями организуются широкомасштабные информационные кампании [66] с целью продвижения добровольного добрачного скрининга, при этом браки между носителями категорически не поощряются.
Эпидемиология
Бета-форма талассемии особенно распространена среди народов Средиземноморья , и это географическое объединение дало ей первоначальное название. [67] Талассемия привела к 25 000 смертей в 2013 году по сравнению с 36 000 смертей в 1990 году. [15]
Заболевание также встречается у населения, живущего в Африке, Америке, а также у народа тару в регионе Тераи в Непале и Индии . [69] Считается, что именно этим объясняется гораздо более низкий уровень заболеваемости и смертности от малярии, [70] что объясняет историческую способность тарусов выживать в районах с тяжелым заражением малярией, в то время как другие не могли этого сделать. Талассемия особенно связана с людьми средиземноморского происхождения, арабами (особенно палестинцами и людьми палестинского происхождения) и азиатами. [71] По оценкам, распространенность составляет 16% среди жителей Кипра , 1% [72] в Таиланде и 3–8% среди населения Бангладеш , Китая , Индии , Малайзии и Пакистана .
По оценкам, примерно 1,5% мирового населения (80–90 миллионов человек) являются носителями β-талассемии. [73] Однако точные данные о показателях носителей во многих группах населения отсутствуют, особенно в развивающихся регионах мира, которые, как известно или ожидаются, будут сильно затронуты. [74] [75] Из-за распространенности заболевания в странах, где о талассемии мало что известно, доступ к правильному лечению и диагностике может быть затруднен. [76] Хотя в развивающихся странах существуют некоторые диагностические и лечебные учреждения, в большинстве случаев они не предоставляются государственными службами и доступны только тем пациентам, которые могут себе это позволить. В целом, более бедные слои населения имеют доступ лишь к ограниченным диагностическим средствам и переливанию крови. В некоторых развивающихся странах практически нет средств для диагностики и лечения талассемии. [76]
Этимология и синоним
Слово талассемия ( / θ æ l ɪ ˈ s iː m i ə / ) происходит от греческого thalassa (θάλασσα), «море», [77] и неолатинского -emia (от греческой составной основы - aimia (-αιμία ), от хайма (αἷμα), «кровь»). [78] Оно было придумано потому, что состояние, называемое «средиземноморской анемией», было впервые описано у людей средиземноморской национальности. «Средиземноморская анемия» была переименована в большую талассемию , когда генетика стала лучше изучена. Слово талассемия впервые было использовано в 1932 году. [67] : 877 [79]
Исследовать
Генная терапия
Генная терапия изучается при талассемии. [80] Процедура включает сбор гемопоэтических стволовых клеток (ГСК) из крови пострадавшего. Затем к ЗКП добавляют ген бета-глобина с использованием лентивирусного вектора . После разрушения костного мозга больного человека дозой химиотерапии (режим миелоаблативного кондиционирования) измененные ЗКП вводятся обратно в организм больного, где они приживляются в костном мозге и размножаются. Это потенциально приводит к постепенному увеличению синтеза гемоглобина А2 во всех последующих развивающихся эритроцитах, что приводит к разрешению анемии. [81]
Хотя одному человеку с бета-талассемией больше не требовалось переливание крови после лечения в рамках научного исследования, по состоянию на 2018 год это лечение не одобрено. [80] [82]
Индукция HbF
Индукция HbF представляет собой попытку реактивировать транскрипцию гена глобина плода. [83] Усилия включают попытку разрушить промотор гена глобина плода. [83]
Рекомендации
^ abcdefgh «Каковы признаки и симптомы талассемии?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 16 сентября 2016 года . Проверено 5 сентября 2016 г.
^ abc «Что вызывает талассемию?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 26 августа 2016 года . Проверено 5 сентября 2016 г.
^ ab «Как диагностируется талассемия?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 16 сентября 2016 года . Проверено 5 сентября 2016 г.
^ abcde «Как лечат талассемию?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 16 сентября 2016 года . Проверено 5 сентября 2016 г.
^ GBD 2015 Заболеваемость и распространенность травм и заболеваний (8 октября 2016 г.). «Глобальная, региональная и национальная заболеваемость, распространенность и годы жизни с инвалидностью по 310 заболеваниям и травмам, 1990–2015 гг.: систематический анализ для исследования глобального бремени болезней, 2015 г.». Ланцет . 388 (10053): 1545–1602. дои : 10.1016/S0140-6736(16)31678-6. ПМК 5055577 . ПМИД 27733282.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ ab GBD 2015 Смертность и причины смерти (8 октября 2016 г.). «Глобальная, региональная и национальная ожидаемая продолжительность жизни, смертность от всех причин и смертность от конкретных причин по 249 причинам смерти, 1980–2015 гг.: систематический анализ для исследования глобального бремени болезней, 2015 г.». Ланцет . 388 (10053): 1459–1544. дои : 10.1016/s0140-6736(16)31012-1. ПМЦ 5388903 . ПМИД 27733281.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ abc «Что такое талассемия?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 26 августа 2016 года . Проверено 5 сентября 2016 г.
^ «Как можно предотвратить талассемию?». НХЛБИ . 3 июля 2012 года. Архивировано из оригинала 16 сентября 2016 года . Проверено 5 сентября 2016 г.
^ «Хелирование железа» . Проверено 15 июля 2020 г.
^ Шах, Сандип; Шет, Радхика; Шах, Камлеш; Патель, Киннари (февраль 2020 г.). «Безопасность и эффективность комбинации талидомида и гидроксимочевины при средней и большой β-талассемии: ретроспективное пилотное исследование». Британский журнал гематологии . 188 (3): e18–e21. дои : 10.1111/bjh.16272 . ISSN 0007-1048. PMID 31710694. S2CID 207940189.
^ Кейхаи, Биджан (2015). «Клинические и гематологические эффекты гидроксимочевины у пациентов с промежуточной β-талассемией». Журнал клинических и диагностических исследований . 9 (10): ОМ01-3. дои : 10.7860/JCDR/2015/14807.6660. ПМК 4625280 . ПМИД 26557561.
^ Мазера, Николетта; Тавеккья, Луиза; Капра, Мариетта; Каццанига, Джованни; Вимеркати, Кьяра; Поцци, Лорена; Бионди, Андреа; Мазера, Джузеппе (2010). «Оптимальный ответ на талидомид у пациента с большой талассемией, резистентного к традиционной терапии». Переливание крови . 8 (1): 63–5. дои : 10.2450/2009.0102-09. ISSN 1723-2007. ПМК 2809513 . ПМИД 20104280.
^ Исследование глобального бремени болезней, 2013 г. (22 августа 2015 г.). «Глобальная, региональная и национальная заболеваемость, распространенность и количество лет жизни с инвалидностью для 301 острого и хронического заболевания и травмы в 188 странах, 1990–2013 гг.: систематический анализ для исследования глобального бремени болезней 2013 г.». Ланцет . 386 (9995): 743–800. дои : 10.1016/s0140-6736(15)60692-4. ПМЦ 4561509 . ПМИД 26063472.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ ab GBD 2013 Смертность и причины смерти (17 декабря 2014 г.). «Глобальная, региональная и национальная смертность от всех причин и по конкретным причинам в разбивке по возрасту и по конкретным причинам по 240 причинам смерти, 1990–2013 гг.: систематический анализ для исследования глобального бремени болезней, 2013 г.». Ланцет . 385 (9963): 117–71. дои : 10.1016/S0140-6736(14)61682-2. hdl : 11655/15525. ПМК 4340604 . ПМИД 25530442.{{cite journal}}: CS1 maint: числовые имена: список авторов ( ссылка )
^ Weatherall, DJ (2015). «Талассемия: нарушения синтеза глобина». Гематология Уильямса (9-е изд.). МакГроу Хилл Профессионал. п. 725. ИСБН9780071833011.
^ Чианчулли П. (октябрь 2008 г.). «Лечение перегрузки железом при талассемии». Педиатр Эндокринол Ред . 6 (Приложение 1): 208–13. ПМИД 19337180.
^ «Талассемия - Симптомы и причины» . Клиника Майо . Архивировано из оригинала 20 ноября 2016 года . Проверено 4 апреля 2017 г.
^ Вогиаци, Мария Г; Маклин, Эрик А; Фунг, Эллен Б; Чунг, Анджела М; Вичинский, Эллиот; Оливьери, Нэнси; Кирби, Мелани; Квятковски, Джанет Л; Каннингем, Мелоди; Холм, Ингрид А; Лейн, Джозеф; Шнайдер, Роберт; Флейшер, Мартин; Грейди, Роберт В.; Петерсон, Чарльз С; Джардина, Патрисия Дж (март 2009 г.). «Заболевания костей при талассемии: частая и все еще нерешенная проблема». Журнал исследований костей и минералов . 24 (3): 543–557. дои : 10.1359/jbmr.080505. ISSN 0884-0431. ПМК 3276604 . ПМИД 18505376.
^ «Симптомы и причины - Увеличение селезенки (спленомегалия) - Клиника Майо» . www.mayoclinic.org . Архивировано из оригинала 19 ноября 2016 года . Проверено 2 февраля 2017 г.
^ Солиман, Ашраф Т; Калра, Санджай; Де Санктис, Винченцо (1 ноября 2014 г.). «Анемия и рост». Индийский журнал эндокринологии и метаболизма . 18 (7): С1–5. дои : 10.4103/2230-8210.145038 . ПМЦ 4266864 . ПМИД 25538873.
^ «Осложнения талассемии». Талассемия . Открытое издательство. Архивировано из оригинала 3 октября 2011 года . Проверено 27 сентября 2011 г.
^ Weatherall DJ. Новая генетика и клиническая практика, Oxford University Press, Оксфорд, 1991.
^ Хьюсман Т.Д. Строение и функции нормальных и аномальных гемоглобинов. В: Клиническая гематология Байера, Хиггс Д.Р., Weatherall DJ (редакторы), WB Saunders, Лондон, 1993. стр.1.
^ Натараджан К., Таунс Т.М., Кутлар А. Нарушения структуры гемоглобина: серповидноклеточная анемия и связанные с ней аномалии. В: Гематология Уильямса, 8-е изд., Каушанский К., Лихтман М.А., Бойтлер Э. и др. (Ред.), McGraw-Hill, 2010. стр. 48.
^ Вай Фэн Лим, Логесваран Мунианди, Элизабет Джордж, Джамила Сатар, Лай Куан Тех и Мэй И Лай (2015) HbF при HbE/β-талассемии: клиническая и лабораторная корреляция, Гематология, 20:6, 349-353, DOI: 10.1179/1607845414Y.0000000203
^ abc Хусейн, Норита; Хеннеман, Лидевий; Кай, Джо; Куреши, Надим (11 октября 2021 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Оценка риска талассемии, серповидно-клеточной анемии, муковисцидоза и болезни Тея-Сакса до зачатия». Кокрейновская база данных систематических обзоров . 2021 (10): CD010849. дои : 10.1002/14651858.CD010849.pub4. ПМЦ 8504980 . ПМИД 34634131.
^ Вамбуа С; Мванги, Табита В.; Корток, Моисей; Уйога, Софи М.; Махария, Алекс В.; Мвачаро, Джедида К.; Уэзералл, Дэвид Дж.; Сноу, Роберт В.; Марш, Кевин; Уильямс, Томас Н. (май 2006 г.). «Влияние α +-талассемии на заболеваемость малярией и другими заболеваниями у детей, живущих на побережье Кении». ПЛОС Медицина . 3 (5): е158. doi : 10.1371/journal.pmed.0030158 . ПМЦ 1435778 . ПМИД 16605300.
^ Тассиопулос С; Дефтереос, Спирос; Константинопулос, Костас; Фармакис, Димитрис; Цирони, Мария; Кириакидис, Михалис; Эссоп, Афанасий (2005). «Дает ли гетерозиготная бета-талассемия защиту от ишемической болезни сердца?». Анналы Нью-Йоркской академии наук . 1054 (1): 467–70. Бибкод : 2005NYASA1054..467T. дои : 10.1196/анналы.1345.068. PMID 16339699. S2CID 71993591.
^ Герберт Л. Манси-младший; Кэмпбелл, Джеймс С. (15 августа 2009 г.). «Альфа и бета-талассемия». Американский семейный врач . 80 (4): 339–344. ПМИД 19678601.
^ «Дельта-бета-талассемия». Сирота . Проверено 16 сентября 2016 г.
^ «HBD - дельта субъединицы гемоглобина» . Сирота . Проверено 17 сентября 2016 г.
^ Торрес Лде С (март 2015 г.). «Гемоглобин D-Пенджаб: происхождение, распространение и лабораторная диагностика». Бразильский обзор гематологии и гемотерапии . 37 (2): 120–126. дои : 10.1016/j.bjhh.2015.02.007. ПМЦ 4382585 . ПМИД 25818823.
^ «Как диагностируется талассемия? - NHLBI, NIH» . www.nhlbi.nih.gov . Архивировано из оригинала 28 июля 2017 года . Проверено 6 сентября 2017 г.
^ Кеохан, Э; Смит, Л; Валенга, Дж (2015). Гематология Родака: клинические принципы и приложения (5-е изд.). Elsevier Науки о здоровье. стр. 467–9. ISBN978-0-323-23906-6.
^ Коттке-Марчант, К; Дэвис, Б. (2012). Лабораторная гематологическая практика (1-е изд.). Джон Уайли и сыновья. п. 569. ИСБН978-1-4443-9857-1.
^ abc Foong, Вай Ченг; Лох, К Хай; Хо, Жаклин Дж; Лау, Дорис СК (13 января 2023 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Индукторы фетального гемоглобина для уменьшения переливания крови при нетрансфузионно-зависимых бета-талассемиях». Кокрейновская база данных систематических обзоров . 2023 (1): CD013767. дои : 10.1002/14651858.CD013767.pub2. ПМЦ 9837847 . ПМИД 36637054.
^ abcd Нойфельд, EJ (2010). «Обновленная информация о хелаторах железа при талассемии». Гематология . 2010 : 451–5. doi : 10.1182/asheducation-2010.1.451 . ПМИД 21239834.
^ Фишер, Шейла А.; Бранскилл, Сьюзен Дж.; Дори, Кэролайн; Чоудхури, Онима; Гудинг, Сара; Робертс, Дэвид Дж. (21 августа 2013 г.). «Оральный деферипрон для хелирования железа у людей с талассемией». Кокрейновская база данных систематических обзоров (8): CD004839. дои : 10.1002/14651858.CD004839.pub3. ISSN 1469-493X. ПМИД 23966105.
^ аб Боллиг, Клаудия; Шелл, Лиза К.; Рюкер, Герта; Аллерт, Роман; Мотшалл, Эдит; Нимейер, Шарлотта М.; Басслер, Дирк; Меерполь, Йорг Дж. (15 августа 2017 г.). «Деферасирокс для борьбы с перегрузкой железом у людей с талассемией». Кокрановская база данных систематических обзоров . 2017 (8): CD007476. дои : 10.1002/14651858.CD007476.pub3. ISSN 1469-493X. ПМК 6483623 . ПМИД 28809446.
^ Садаф, Алина; Хасан, Бабар; Дас, Джай К; Колан, Стивен; Алви, Наджвин (12 июля 2018 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Блокаторы кальциевых каналов для профилактики кардиомиопатии из-за перегрузки железом у людей с трансфузионно-зависимой бета-талассемией». Кокрейновская база данных систематических обзоров . 2018 (7): CD011626. дои : 10.1002/14651858.CD011626.pub2. ПМК 6513605 . ПМИД 29998494.
^ Нгим, CF; Лай, Нью-Мексико; Хонг, JY; Тан, СЛ; Рамадас, А; Мутукумарасами, П; Тонг, МК (28 мая 2020 г.). «Терапия гормоном роста для людей с талассемией». Кокрановская база данных систематических обзоров . 2020 (5): CD012284. дои : 10.1002/14651858.CD012284.pub3. ПМЦ 7387677 . ПМИД 32463488.
^ Аб Газиев, Дж; Лукарелли, Дж. (июнь 2011 г.). «Трансплантация гемопоэтических стволовых клеток при талассемии». Современные исследования и терапия стволовыми клетками . 6 (2): 162–9. дои : 10.2174/157488811795495413. ПМИД 21190532.
^ Саблофф, М; Чанди, М; Ван, З; Логан, БР; Гавамзаде, А; Лизать; Ирфан, С.М.; Бредесон, Китай; и другие. (2011). «Трансплантация HLA-совместимого брата и сестры костного мозга при большой β-талассемии». Кровь . 117 (5): 1745–50. doi : 10.1182/blood-2010-09-306829. ПМК 3056598 . ПМИД 21119108.
^ Шарма, Акшай; Джаганнатх, Ванита А.; Пури, Латика (21 апреля 2021 г.). «Трансплантация гемопоэтических стволовых клеток людям с β-талассемией». Кокрановская база данных систематических обзоров . 2021 (4): CD008708. дои : 10.1002/14651858.CD008708.pub5. ISSN 1469-493X. ПМК 8078520 . ПМИД 33880750.
^ Фишер, Шейла А; Катлер, Энтони; Дори, Кэролайн; Бранскилл, Сьюзен Дж; Стэнворт, Саймон Дж; Наваррете, Кристина; Гердлстоун, Джон (30 января 2019 г.). Кокрейновская группа по гематологическим злокачественным новообразованиям (ред.). «Мезенхимальные стромальные клетки как лечение или профилактика острой или хронической реакции трансплантат против хозяина у реципиентов трансплантата гемопоэтических стволовых клеток (ТГСК) с гематологическими заболеваниями». Кокрейновская база данных систематических обзоров . 1 (1): CD009768. дои : 10.1002/14651858.CD009768.pub2. ПМК 6353308 . ПМИД 30697701.
^ Содани, П; Исгро, А; Газиев Дж.; Пачиарони, К; Марциали, М; Симона, доктор медицины; Роведа, А; Де Анджелис, Дж; и другие. (2011). «Трансплантация hla-гаплоидентичных стволовых клеток с обеднением Т-клеток молодым пациентам с талассемией». Педиатрические отчеты . 3 (Дополнение 2): e13. doi :10.4081/pr.2011.s2.e13. ПМК 3206538 . ПМИД 22053275.
^ Ансари, Сакиб Х; Ласси, Зохра С; Ховаджа, Салима М; Адиль, Сайед Омайр; Шамси, Тахир С. (16 марта 2019 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Гидроксимочевина (гидроксикарбамид) при трансфузионно-зависимой β-талассемии». Кокрейновская база данных систематических обзоров . 3 (3): CD012064. дои : 10.1002/14651858.CD012064.pub2. ПМК 6421980 . ПМИД 30882896.
^ Ке Мон Мин Све (2013). «Пищевые добавки цинка для лечения талассемии и серповидноклеточной анемии». Кокрейновская база данных систематических обзоров . 2013 (6): CD009415. дои : 10.1002/14651858.CD009415.pub2. ПМЦ 9964104 . ПМИД 23807756.
^ Мулимани, Прити; Абас, Адинегара Б.Л.; Карант, Лакшминараян; Коломбатти, Рафаэлла; Кулкарни, Пална (2 августа 2019 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Лечение стоматологических и ортодонтических осложнений при талассемии». Кокрейновская база данных систематических обзоров . 8 (8): CD012969. дои : 10.1002/14651858.CD012969.pub2. ПМК 6699676 . ПМИД 31425614.
^ аб Бхардвадж, Амит; Шве, Ке Мон Мин; Синха, Нирмал К.; Осункво, Ифейинва (10 марта 2016 г.). «Лечение остеопороза у людей с β-талассемией». Кокрановская база данных систематических обзоров . 3 : CD010429. дои : 10.1002/14651858.CD010429.pub2. ISSN 1469-493X. ПМИД 26964506.
^ abc Мулимани, Прити; Абас, Адинегара Б.Л.; Карант, Лакшминараян; Коломбатти, Рафаэлла; Кулкарни, Пална (2 февраля 2023 г.). Кокрейновская группа по муковисцидозу и генетическим заболеваниям (ред.). «Лечение стоматологических и ортодонтических осложнений при талассемии». Кокрейновская база данных систематических обзоров . 2023 (2): CD012969. дои : 10.1002/14651858.CD012969.pub3. ПМЦ 9893875 . ПМИД 36732291.
^ Бердик, Колорадо; Нтайос, Г.; Ратод, Д. (март 2009 г.). «Разделение признаков талассемии и дефицита железа простым осмотром». Являюсь. Дж. Клин. Патол . 131 (3): 444, ответ автора 445. doi : 10.1309/AJCPC09VRAXEASMH . ПМИД 19228649.
^ «Скрининг носителей в эпоху геномной медицины - ACOG». www.acog.org . Архивировано из оригинала 25 февраля 2017 года . Проверено 24 февраля 2017 г.
^ Люнг, Т.Н.; Лау ТК; Чунг ТХ (апрель 2005 г.). «Скрининг талассемии во время беременности». Современное мнение в акушерстве и гинекологии . 17 (2): 129–34. doi : 10.1097/01.gco.0000162180.22984.a3. PMID 15758603. S2CID 41877258.
^ Лукопулос, Д. (октябрь 2011 г.). «Гемоглобинопатии в Греции: программа профилактики за последние 35 лет». Индийский журнал медицинских исследований . 134 (4): 572–6. ПМЦ 3237258 . ПМИД 22089622.
^ Самават А, Модель Б (ноябрь 2004 г.). «Иранская национальная программа скрининга талассемии». BMJ (Клинические исследования под ред.) . 329 (7475): 1134–7. дои : 10.1136/bmj.329.7475.1134. ПМК 527686 . ПМИД 15539666.
↑ Петру, Мэри (1 января 2010 г.). «Скрининг бета-талассемии». Индийский журнал генетики человека . 16 (1): 1–5. doi : 10.4103/0971-6866.64934 (неактивен 7 февраля 2024 г.). ПМЦ 2927788 . ПМИД 20838484.{{cite journal}}: CS1 maint: DOI неактивен по состоянию на февраль 2024 г. ( ссылка )[ постоянная мертвая ссылка ]
^ аб Грир, Джон П.; Арбер, Дэниел А.; Глэйдер, Бертиль; Лист, Алан Ф.; Минс-младший, Роберт Т.; Параскевас, Фриксос; Роджерс, Джордж М.; Ферстер, Джон (2013). Клиническая гематология Винтроба . Уолтерс Клювер, Липпинкотт Уильямс и Уилкинс Хелс. ISBN9781451172683.
^ Вахид, Фазила; Фистер, Коллин; Авофесо, АвоНийи; Стэнли, Дэвид (июль 2016 г.). «Скрининг носительства бета-талассемии на Мальдивах: восприятие родителей больных детей, которые не принимали участие в скрининге, и его последствия». Журнал общественной генетики . 7 (3): 243–253. дои : 10.1007/s12687-016-0273-5. ПМК 4960032 . ПМИД 27393346.
^ Модиано, Г.; Морпурго, Дж; Терренато, Л; Новеллетто, А; Ди Риенцо, А; Коломбо, Б; Пурпура, М; Мариани, М; и другие. (1991). «Защита от заболеваемости малярией: почти фиксация гена α-талассемии в непальском населении». Американский журнал генетики человека . 48 (2): 390–7. ПМК 1683029 . ПМИД 1990845.
^ Терренато, Л; Шреста, С; Диксит, Калифорния; Луццато, Л; Модиано, Дж; Морпурго, Дж; Арезе, П. (февраль 1988 г.). «Снижение заболеваемости малярией среди населения Тару по сравнению с симпатрическим населением Непала». Анналы тропической медицины и паразитологии . 82 (1): 1–11. дои : 10.1080/00034983.1988.11812202. ПМИД 3041928.
^ Э. Гольян, Патология, 2-е изд. Mosby Elsevier, серия Rapid Review. [ нужна страница ]
^ «Талассемия» (на тайском языке). Департамент медицинских наук. Сентябрь 2011 г. Архивировано из оригинала 25 сентября 2011 г.
^ Вичинский, Эллиот П. (1 ноября 2005 г.). «Изменение моделей талассемии во всем мире». Анналы Нью-Йоркской академии наук . 1054 (1): 18–24. Бибкод : 2005NYASA1054...18V. дои : 10.1196/анналы.1345.003. ISSN 1749-6632. PMID 16339647. S2CID 26329509.
^ ab Weatherall, Дэвид Дж. (ноябрь 2005 г.). «Основной доклад: Проблема талассемии для развивающихся стран». Анналы Нью-Йоркской академии наук . 1054 (1): 11–17. Бибкод : 2005NYASA1054...11W. дои : 10.1196/анналы.1345.002. PMID 16339646. S2CID 45770891.
^ Уиппл, GH; Брэдфорд, Висконсин (1932). «Расовая или семейная анемия детей, связанная с фундаментальными нарушениями метаболизма костей и пигментов (Кули-Фон Якш)». Американский журнал болезней детей . 44 : 336–365. doi : 10.1001/archpedi.1932.01950090074009.
↑ Биффи, А. (19 апреля 2018 г.). «Генная терапия как вариант лечения β-талассемии». Медицинский журнал Новой Англии . 378 (16): 1551–1552. дои : 10.1056/NEJMe1802169. ПМИД 29669229.
^ Лидонничи, MR; Феррари, Г (май 2018 г.). «Генная терапия и стратегии редактирования генов при гемоглобинопатиях». Клетки крови, молекулы и болезни . 70 : 87–101. дои : 10.1016/j.bcmd.2017.12.001. ПМИД 29336892.
^ аб Винерт, Б; Мартин, GE; Фаннелл, APW; Куинлан, КГР; Кроссли, М. (1 октября 2018 г.). «Ген пробуждения сна: реактивация фетального глобина при β-гемоглобинопатиях». Тенденции в генетике . 34 (12): 927–940. дои :10.1016/j.tig.2018.09.004. PMID 30287096. S2CID 52921922.