Амилоидоз — это группа заболеваний, при которых в тканях накапливаются аномальные белки , известные как амилоидные фибриллы . [4] Существует несколько неспецифических и расплывчатых признаков и симптомов, связанных с амилоидозом. [5] К ним относятся утомляемость, периферические отеки , потеря веса, одышка, сердцебиение и слабость при стоянии . [5] При AL-амилоидозе специфические показатели могут включать увеличение языка и периорбитальную пурпуру . [5] При ATTR-амилоидозе дикого типа несердечные симптомы включают: двусторонний синдром запястного канала , стеноз поясничного отдела позвоночника , разрыв сухожилия двуглавой мышцы , невропатию мелких волокон и вегетативную дисфункцию . [5]
Существует около 36 различных типов амилоидоза, каждый из которых возникает из-за неправильного сворачивания определенного белка . [6] Среди этих 36 белков 19 сгруппированы в локализованные формы , 14 сгруппированы как системные формы , а три белка можно идентифицировать как любую из них. [6] Эти белки могут стать нерегулярными из-за генетических эффектов, а также из-за приобретенных факторов окружающей среды . [6] Четырьмя наиболее распространенными типами системного амилоидоза являются легкие цепи (AL) , воспаление ( AA ), диализное заболевание (Aβ 2 M), а также наследственный и старческий ( ATTR и транстиретиновый амилоид дикого типа [7] ). [2]
Диагноз можно заподозрить, когда в моче обнаруживается белок , наблюдается увеличение органов или обнаруживаются проблемы с множественными периферическими нервами , и неясно, почему. [2] Диагноз подтверждается биопсией тканей . [2] Из-за разнообразия проявлений диагноз часто может занять некоторое время. [3]
Лечение направлено на уменьшение количества вовлеченного белка. [2] Иногда этого можно достичь путем определения и лечения основной причины. [2] AL-амилоидоз встречается примерно у 3–13 на миллион человек в год, а АА-амилоидоз – примерно у двух на миллион человек в год. [2] Обычный возраст начала этих двух типов составляет от 55 до 60 лет. [2] Без лечения ожидаемая продолжительность жизни составляет от шести месяцев до четырех лет. [2] В развитых странах примерно один случай смерти из 1000 приходится на системный амилоидоз. [3] Амилоидоз описывается как минимум с 1639 года. [2]
Проявления амилоидоза широки и зависят от места накопления амилоида. Почки и сердце являются наиболее часто поражаемыми органами.
Отложение амилоида в почках часто затрагивает капилляры клубочков и мезангиальные области , влияя на способность органа фильтровать и выводить отходы, а также удерживать белки плазмы . [8] Это может привести к повышению уровня белка в моче ( протеинурии ) и нефротическому синдрому . [8] Несколько типов амилоидоза, включая типы AL и AA, связаны с нефротическим синдромом . [9] Примерно у 20% и 40–60% людей с АЛ и АА-амилоидозом соответственно развивается терминальная стадия заболевания почек, требующая диализа . [9]
Отложение амилоида в сердце может вызвать как диастолическую, так и систолическую сердечную недостаточность . Могут присутствовать изменения ЭКГ , демонстрирующие низкий вольтаж и нарушения проводимости, такие как атриовентрикулярная блокада или дисфункция синусового узла . [ нужна медицинская ссылка ] На эхокардиографии сердце показывает ограничительный характер наполнения, с нормальной или слегка сниженной систолической функцией. [10] АА-амилоидоз обычно поражает сердце. [11] Сердечный амилоидоз может проявляться симптомами сердечной недостаточности, включая одышку, усталость и отеки. [12] По мере прогрессирования сердечного амилоидоза отложение амилоида может повлиять на способность сердца перекачивать и наполнять кровь, а также на его способность поддерживать нормальный ритм, что приводит к ухудшению функции сердца и снижению качества жизни людей. [12]
У людей с амилоидозом может наблюдаться поражение центральной нервной системы [13] наряду с поражением периферической нервной системы, что приводит к сенсорным и вегетативным нейропатиям. Сенсорная нейропатия развивается симметрично и прогрессирует в дистальном и проксимальном направлении. Автономная нейропатия может проявляться как ортостатическая гипотензия , но может проявляться более постепенно с неспецифическими желудочно-кишечными симптомами, такими как запор, тошнота или раннее насыщение. [10] Амилоидоз центральной нервной системы может иметь более тяжелые и системные проявления, которые могут включать опасные для жизни аритмии, сердечную недостаточность, недостаточность питания, инфекцию или смерть. [14]
Невропатическая картина может зависеть от этиологии амилоидоза. [14] Люди с амилоидозом могут испытывать дисфункцию различных систем органов в зависимости от локализации и степени поражения нервной системы. [8] Например, периферическая нейропатия может вызывать эректильную дисфункцию, недержание и запор, дисфункцию зрачков и потерю чувствительности в зависимости от распределения амилоидоза по различным периферическим нервам. [14]
Накопление амилоидных белков в желудочно-кишечной системе может быть обусловлено широким спектром амилоидных нарушений и иметь различную картину в зависимости от степени поражения органов. [15] Потенциальные симптомы включают потерю веса, диарею, боль в животе, изжогу (желудочный рефлюкс) и желудочно-кишечное кровотечение. [15] Амилоидоз может также поражать дополнительные органы пищеварения, включая печень, и проявляться желтухой, жирным стулом, анорексией, скоплением жидкости в брюшной полости и увеличением селезенки. [15]
Накопление амилоидных белков в печени может привести к повышению уровня сывороточных аминотрансфераз и щелочной фосфатазы , двух биомаркеров поражения печени, что наблюдается примерно у трети людей. [11] Часто наблюдается увеличение печени . Напротив, увеличение селезенки встречается редко и встречается у 5% людей. [10] Дисфункция селезенки, приводящая к наличию телец Хауэлла-Джолли в мазке крови, встречается у 24% людей с амилоидозом. [10] Мальабсорбция наблюдается в 8,5% случаев АЛ-амилоидоза и в 2,4% случаев АА-амилоидоза. Один из предполагаемых механизмов наблюдаемой мальабсорбции заключается в том, что отложения амилоида на кончиках кишечных ворсинок (пальцеобразные выступы, которые увеличивают площадь кишечника, доступную для всасывания пищи) начинают разрушать функциональность ворсинок, создавая картину, подобную слитку . [11]
Инфильтрации могут подвергаться как щитовидная железа , так и надпочечники . По оценкам, 10–20% людей с амилоидозом страдают гипотиреозом . Инфильтрацию надпочечников оценить труднее, поскольку ее симптомы в виде ортостатической гипотензии и низкой концентрации натрия в крови могут быть связаны с автономной нейропатией и сердечной недостаточностью. [10]
«Отложения амилоида возникают в поджелудочной железе у людей, страдающих сахарным диабетом , хотя неизвестно, имеет ли это функциональное значение. Основным компонентом амилоида поджелудочной железы является пептид из 37 аминокислотных остатков, известный как островковый амилоидный полипептид или «амилин». Он хранится вместе с инсулином в секреторных гранулах бета-клеток и секретируется совместно с инсулином». ( Фармакология Рэнга и Дейла, 2015 г. )
Амилоидные белки откладываются чаще всего в коленях, затем в руках, запястьях, локтях, бедрах и лодыжках, вызывая боль в суставах. [16] У мужчин пожилого возраста (>80 лет) существует значительный риск отложения амилоида транстиретина дикого типа в синовиальной ткани коленного сустава, но преимущественно в пожилом возрасте отложение транстиретина дикого типа наблюдается в желудочках сердца. Отложения ATTR были обнаружены в желтой связке пациентов, перенесших операцию по поводу стеноза поясничного отдела позвоночника . [17]
При бета-2-микроглобулиновом амилоидозе мужчины имеют высокий риск развития синдрома запястного канала . [18] Амилоидоз Aβ2MG (амилоидоз, связанный с гемодиализом) имеет тенденцию откладываться в синовиальной ткани, вызывая хроническое воспаление синовиальной ткани в коленных, тазобедренных, плечевых и межфаланговых суставах. [18] Отложение легких цепей амилоида в плечевом суставе вызывает увеличение плеч, также известное как « признак подплечника ». [18] Отложения легких цепей амилоида также могут вызывать двусторонний симметричный полиартрит. [18]
Отложение амилоидных белков в костном мозге без возникновения дискразий плазматических клеток называется амилоидомой. Чаще всего встречается в шейных, поясничных и крестцовых позвонках. У пострадавших могут наблюдаться боли в костях из-за лизиса костей, поясничного парапареза и различных неврологических симптомов. Переломы позвонков также распространены. [18]
Редким развитием является амилоидная пурпура , склонность к кровотечениям с синяками вокруг глаз, называемая «енотовидными глазами». Амилоидная пурпура вызвана отложением амилоида в кровеносных сосудах и снижением активности тромбина и фактора X , двух белков свертывания крови, которые теряют свою функцию после связывания с амилоидом. [10]
Отложения амилоида в тканях могут вызывать увеличение структур. Двадцать процентов людей с AL-амилоидозом имеют увеличенный язык , что может привести к обструктивному апноэ во сне , затруднению глотания и изменению вкуса. [11] Увеличение языка не происходит при амилоидозе ATTR или АА. [10] Отложение амилоида в горле может вызвать охриплость голоса. [10]
Амилоидозы можно рассматривать как заболевания неправильного сворачивания белков . [19] [20] Подавляющее большинство белков, которые, как было обнаружено, образуют амилоидные отложения, являются секретируемыми белками , поэтому неправильное сворачивание и образование амилоида происходит вне клеток, во внеклеточном пространстве. [19] Из 37 белков, которые на данный момент идентифицированы как уязвимые к образованию амилоида, только четыре являются цитозольными . [19] Большинство амилоидобразующих белков относительно малы, но в остальном в настоящее время нет доказательств структурного или функционального сходства между белками, которые, как известно, образуют амилоиды, связанные с заболеваниями. [19] Треть случаев амилоидной болезни является наследственной, и в этом случае заболевание обычно начинается в раннем возрасте. [19] Половина заболеваний, связанных с амилоидом, являются спорадическими и имеют поздний возраст начала – в этих случаях агрегация белка может быть связана с возрастным снижением регуляции белка. Некоторые методы лечения связаны с амилоидной болезнью, но это случается редко. [19]
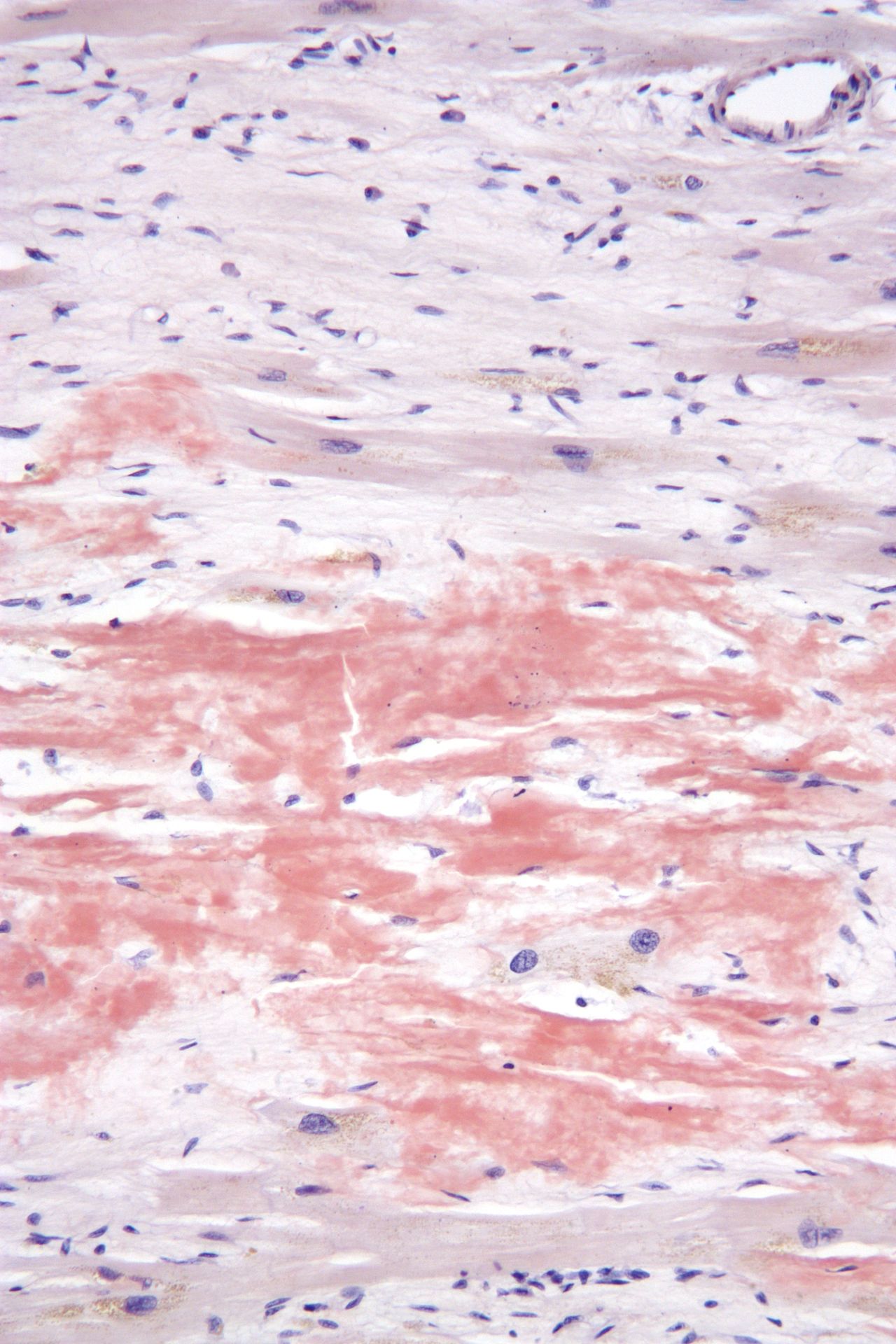
Белки, образующие амилоид, агрегируют в характерные фибриллярные формы со структурой бета-листа . [19] [20] Бета-листная форма амилоида устойчива к протеолизу , то есть не поддается деградации или расщеплению. [5] В результате амилоид откладывается во внеклеточном пространстве организма. [5] Считается, что процесс формирования амилоидных фибрилл имеет промежуточные олигомерные формы. И олигомеры, и амилоидные фибриллы могут быть токсичными для клеток и нарушать правильное функционирование органов. [21] Относительная значимость различных видов агрегаций может зависеть от задействованного белка и пораженной системы органов. [20]
Диагностика амилоидоза обычно требует биопсии ткани. [2] Биопсия оценивается на наличие характерных отложений амилоида. Ткань обрабатывается различными морилками . Наиболее полезным красителем для диагностики амилоида является конго красный , который в сочетании с поляризованным светом придает амилоидным белкам при микроскопии яблочно-зеленый цвет . Также можно использовать краситель тиофлавином Т. [22] Также используется ряд методов визуализации, таких как сканирование PYP Nuclear Medicine, сканирование DPD или сканирование SAP . [23]
Образец ткани можно подвергнуть биопсии или получить непосредственно из пораженного внутреннего органа, но местом биопсии первой линии является подкожный брюшной жир , известный как «биопсия жировой подушечки», из-за простоты его получения. [24] [25] Биопсия брюшного жира не является полностью чувствительной и может дать ложноотрицательный результат, что означает, что отрицательный результат не исключает диагноз амилоидоза. [24] [25] Однако прямая биопсия пораженного органа может оказаться ненужной, поскольку можно использовать и другие менее инвазивные методы биопсии, в том числе биопсию слизистой прямой кишки, слюнной железы, губы или костного мозга, которые позволяют поставить диагноз в более ранние сроки. до 85% людей. [24]
При отложении амилоида в суставах будет наблюдаться снижение сигнала как на Т1, так и на Т2-взвешенных МРТ-изображениях . [16] При амилоидоме будет низкий сигнал Т1 при инъекции гадолиния и низкий сигнал Т2. [18]
Тип амилоидного белка можно определить различными способами: обнаружением аномальных белков в кровотоке (при электрофорезе белков или определении легкой цепи); связывание определенных антител с амилоидом, обнаруженным в ткани (иммуногистохимия); или экстракция белка и идентификация его отдельных аминокислот . [22] Иммуногистохимия позволяет выявить АА-амилоидоз в большинстве случаев, но может пропустить многие случаи AL-амилоидоза. [11] Лазерная микродиссекция с масс-спектрометрией является наиболее надежным методом выявления различных форм амилоидоза. [26]
Ранее АЛ считался наиболее распространенной формой амилоидоза, и диагностика часто начинается с поиска дискразии плазматических клеток , В-клеток памяти, продуцирующих аберрантные иммуноглобулины или части иммуноглобулинов. Иммунофиксационный электрофорез мочи или сыворотки дает положительный результат у 90% людей с AL-амилоидозом. [10] Иммунофиксационный электрофорез более чувствителен, чем обычный электрофорез, но может быть доступен не во всех центрах. В качестве альтернативы можно провести иммуногистохимическое окрашивание биопсии костного мозга в поисках доминантных плазматических клеток у людей с высоким клиническим подозрением на AL-амилоидоз, но отрицательным электрофорезом. [10]
В настоящее время ATTR считается наиболее распространенной формой амилоидоза. Это может быть либо возрастное заболевание при ATTR дикого типа (ATTRv), либо семейный транстиретин-ассоциированный амилоидоз, подозревается у людей с семейным анамнезом идиопатических нейропатий или сердечной недостаточности, у которых отсутствуют признаки дискразии плазматических клеток. ATTR можно идентифицировать с помощью изоэлектрического фокусирования , которое отделяет мутировавшие формы транстиретина. Результаты могут быть подтверждены генетическим тестированием для поиска конкретных известных мутаций транстиретина, которые предрасполагают к амилоидозу. [10]
АА подозревается на основании клинических данных у лиц с длительными инфекциями или воспалительными заболеваниями. АК можно идентифицировать с помощью иммуногистохимического окрашивания. [10]
Исторические системы классификации основывались на клинических факторах. До начала 1970-х годов преобладала идея единого амилоидного вещества. Были предложены различные системы описательной классификации, основанные на распределении амилоидных отложений в органах и клинических данных. Большинство систем классификации включали первичный (т. е. идиопатический ) амилоидоз, при котором не было выявлено сопутствующего клинического состояния, и вторичный амилоидоз (т. е. вторичный по отношению к хроническим воспалительным состояниям). Некоторые системы классификации включали миеломно-ассоциированный, семейный и локализованный амилоидоз. [ нужна цитата ]
Современная эра классификации амилоидоза началась в конце 1960-х годов с разработки методов, позволяющих сделать амилоидные фибриллы растворимыми. Эти методы позволили ученым изучить химические свойства амилоидов. [ нужна медицинская ссылка ] Описательные термины, такие как первичный амилоидоз, вторичный амилоидоз и другие (например, старческий амилоидоз), которые не основаны на причине, дают мало полезной информации и больше не рекомендуются.
Современная классификация амилоидных заболеваний имеет тенденцию использовать аббревиатуру белка, который образует большую часть отложений, с префиксом буквы А. Например, амилоидоз, вызванный транстиретином, называется «ATTR». [ нужна медицинская ссылка ] Характер отложения варьируется у разных людей, но почти всегда состоит только из одного амилоидогенного белка. Отложение может быть системным (затрагивающим множество различных систем органов) или органоспецифичным. Многие амилоидозы передаются по наследству вследствие мутаций в белке-предшественнике. [ нужна медицинская ссылка ]
Другие формы возникают из-за различных заболеваний, вызывающих избыточное или аномальное производство белка, например, из-за перепроизводства легких цепей иммуноглобулина (так называемого AL-амилоидоза ) или постоянного перепроизводства белков острой фазы при хроническом воспалении (что может привести к АА-амилоидозу ). [ нужна медицинская ссылка ]
На данный момент идентифицировано около 60 амилоидных белков. [27] Из них по крайней мере 36 были связаны с болезнями человека. [28]
Все белки амилоидных фибрилл начинаются с буквы «А», за которой следует белковый суффикс (и любая применимая спецификация). Ниже приведен список белков амилоидных фибрилл, обнаруженных у человека: [29]
Более старый клинический метод классификации относится к амилоидозам как к системным или локализованным:
Другая классификация – первичная или вторичная. [ нужна медицинская ссылка ]
Кроме того, в зависимости от тканей, в которых он откладывается, его разделяют на мезенхимальный (органы, происходящие из мезодермы ) или паренхиматозный (органы, происходящие из эктодермы или энтодермы ). [ нужна медицинская ссылка ]
Лечение зависит от типа амилоидоза. Лечение высокими дозами мелфалана , химиотерапевтического агента, с последующей трансплантацией стволовых клеток показало многообещающие результаты в ранних исследованиях и рекомендуется при I и II стадиях AL-амилоидоза. [26] Однако только 20–25% людей имеют право на трансплантацию стволовых клеток. Химиотерапевтическое лечение, включая циклофосфамид-бортезомиб-дексаметазон, в настоящее время является рекомендуемым вариантом лечения для людей с AL-амилоидозом, которым не показана трансплантация. [5]
При АА симптомы могут улучшиться, если лечить основное заболевание. У людей с воспалением, вызванным АА-амилоидозом, ингибиторы фактора некроза опухоли (ФНО)-альфа, такие как инфликсимаб и этанерцепт , используются в среднем в течение 20 месяцев. Если ингибиторы ФНО-альфа не эффективны, можно рассмотреть возможность применения ингибиторов интерлейкина-1 (например, анакинры , канакинумаба , рилонацепта ) и ингибиторов интерлейкина-6 (например, тоцилизумаба). [31]
Лечение амилоидоза ATTR будет зависеть от его классификации как дикого типа или варианта. [5] Оба заболевания можно лечить тафамидисом , малотоксичным пероральным препаратом, который предотвращает дестабилизацию правильно свернутого белка. [5] Исследования показали, что тафамидис снижает смертность и количество госпитализаций из-за сердечной недостаточности . [5] Ранее при вариантном амилоидозе ATTR трансплантация печени была единственным эффективным методом лечения. [5] Новые методы лечения включают дифлунисал , инотерсен и патисиран .
Дифлунисал связывается с неправильно свернутым мутантным белком TTR, предотвращая его накопление, подобно тому, как действует тафамидис. Доказательства низкой определенности указывают на то, что он смягчает ухудшение периферической нейропатии и инвалидность из-за прогрессирования заболевания. [32]
Инотерсен блокирует экспрессию генов как дикого типа, так и мутантного TTR, уменьшая количество предшественников амилоида. Доказательства умеренной определенности свидетельствуют о том, что он смягчает ухудшение периферической нейропатии. Долгосрочная эффективность и безопасность использования инотерсена у людей с мутантным амилоидозом, связанным с TTR, все еще оценивается в III фазе клинических исследований, начиная с 2021 года. И дифлунизал, и инотерсен могут также смягчать снижение качества жизни, хотя доказательства ибо этот эффект неясен. [32] Для людей с сердечным ATTR эффект использования инотерсена неубедителен и требует дальнейшего изучения. [33] В 2018 году Европейское агентство по лекарственным средствам одобрило инотерсен для лечения полинейропатии у взрослых с наследственным транстиретиновым амилоидозом. [34] С тех пор он был одобрен для использования в Канаде, Европейском Союзе и США. [35]
Патисиран действует аналогично инотерсену. Доказательства средней степени достоверности свидетельствуют о том, что патисиран смягчает ухудшение периферической нейропатии и инвалидность вследствие прогрессирования заболевания. Кроме того, данные низкой определенности свидетельствуют о том, что патисиран смягчает снижение качества жизни и немного снижает частоту нежелательных явлений по сравнению с плацебо. Нет никаких доказательств влияния на уровень смертности. [32] Обзор ранних данных по использованию патисирана у людей с вариантным сердечным ATTR позволяет предположить, что он может снизить смертность и количество госпитализаций, однако этот вопрос все еще изучается и требует дальнейшего изучения. [33] В 2018 году NICE в Великобритании не рекомендовал патисиран при наследственном транстиретин-ассоциированном амилоидозе. [36] Однако по состоянию на июль 2019 года проводится дальнейшая проверка. [37] Однако в США он был одобрен для такого использования. [38]
Роль инотерсена и патисирана в сердечном амилоидозе ATTR все еще исследуется. [5]
В 2021 году в клиническом исследовании с использованием метода редактирования генов CRISPR у нескольких участников, получавших патисиран, наблюдалось «падение уровня TTR на 80–96%, что на уровне или лучше, чем в среднем 81%». [39]
Вутрисиран был одобрен Управлением по контролю за продуктами и лекарствами США (FDA) в июне 2022 года для лечения полиневропатии наследственного транстиретин-опосредованного (hATTR) амилоидоза у взрослых. [40]
Людей, страдающих амилоидозом, поддерживают такие организации, как Консорциум по исследованию амилоидоза, Фонд амилоидоза, Группы поддержки амилоидоза и Австралийская сеть по амилоидозу. [41] [42]
Прогноз варьируется в зависимости от типа амилоидоза и пораженной системы органов. Прогноз при нелеченом АЛ-амилоидозе сердца плохой, медиана выживаемости составляет шесть месяцев. [43] Более конкретно, AL-амилоидоз можно классифицировать как стадию I, II или III на основании сердечных биомаркеров, таких как Nt-proBNP и сердечный тропонин. [44] Выживаемость снижается с увеличением стадии, но недавние достижения в лечении улучшили медианную выживаемость для стадий I, II и III до 91,2, 60 и 7 месяцев соответственно. [44]
Исходы у человека с АА-амилоидозом зависят от основного заболевания, пораженного органа (органов) и коррелируют с концентрацией сывороточного белка амилоида А. [5]
Люди с ATTR, мутантным ATTR и ATTR дикого типа имеют лучший прогноз по сравнению с людьми с AL и могут прожить более десяти лет. [10] [45] Время выживания не связано с полом или возрастом, однако некоторые показатели снижения функции сердца связаны с более коротким временем выживания. [45]
Старческий системный амилоидоз был признан основной причиной смерти 70% людей старше 110 лет , прошедших аутопсию . [46] [47]
По оценкам, общая распространенность амилоидоза составляет 30 на 100 000 человек, причем тремя наиболее распространенными формами являются AL, ATTR и AA. [48] Средний возраст на момент постановки диагноза составляет 64 года. [11]
АЛ имеет самую высокую заболеваемость – примерно 12 случаев на миллион человек в год, а распространенность – от 30 000 до 45 000 случаев в США и Европейском Союзе. [48] [5]
АА-амилоидоз является наиболее распространенной формой в развивающихся странах и может осложнять длительные инфекции туберкулезом , остеомиелитом и бронхоэктазами . АА-амилоидоз вызывается увеличением внеклеточного отложения сывороточного белка амилоида А (SAA). Уровни белка SAA могут повышаться как прямым, так и косвенным образом, в результате инфекции, воспаления и злокачественных новообразований. [49] Наиболее распространенными причинами АА-амилоидоза на Западе являются ревматоидный артрит, воспалительные заболевания кишечника, псориаз и семейная средиземноморская лихорадка . [10]
У людей, находящихся на длительном гемодиализе (14–15 лет), амилоидоз может развиться из-за накопления легких цепей комплекса HLA 1, который в норме отфильтровывается почками. [11]
Транстиретиновый (ATTR) амилоидоз дикого типа обнаруживается у четверти пожилых людей при вскрытии. [50] ATTR обнаруживается у 13–19% людей, страдающих сердечной недостаточностью с сохраненной фракцией выброса, что делает его очень распространенной формой системного амилоидоза. [51]
Лечение нейропатии , связанной с ATTR, включает использование TTR-специфичных олигонуклеотидов в форме малой интерферирующей РНК (патисиран) или антисмыслового инотерсена [52] , первый из которых недавно получил одобрение FDA. [53] Исследования по лечению амилоидоза ATTR сравнивали трансплантацию печени, пероральные препараты, которые стабилизируют неправильно сворачивающийся белок (включая тафамидис и дифлунизал), и новые терапевтические агенты, которые все еще исследуются (включая патисиран). [54] Согласно имеющимся исследованиям, трансплантация печени остается наиболее эффективным вариантом лечения распространенного ATTR-амилоидоза, препараты, стабилизирующие белок, могут замедлить прогрессирование заболевания, но их недостаточно, чтобы оправдать отсрочку трансплантации печени, а новые препараты, такие как патисиран, требуют дополнительных исследований. [54]