.jpg/1280px-Clinical_Trial_Participant_Receives_Injection_(34033294061).jpg)
Клинические испытания — это перспективные биомедицинские или поведенческие исследования на людях, призванные ответить на конкретные вопросы о биомедицинских или поведенческих вмешательствах, включая новые методы лечения (такие как новые вакцины , лекарства , диетические предпочтения , диетические добавки и медицинские устройства ) и известные вмешательства, которые требуют дальнейшего изучения и сравнения. Клинические испытания собирают данные о дозировке, безопасности и эффективности. [1] [2] Они проводятся только после получения одобрения органа здравоохранения/этического комитета в стране, где запрашивается одобрение терапии. Эти органы несут ответственность за проверку соотношения риска и пользы испытания — их одобрение не означает, что терапия «безопасна» или эффективна, а только то, что испытание может быть проведено.
В зависимости от типа продукта и стадии разработки исследователи изначально набирают добровольцев или пациентов в небольшие пилотные исследования , а затем проводят все более масштабные сравнительные исследования. Клинические испытания могут различаться по размеру и стоимости, и они могут включать один исследовательский центр или несколько центров , в одной стране или в нескольких странах. Дизайн клинического исследования направлен на обеспечение научной обоснованности и воспроизводимости результатов.
Расходы на клинические испытания могут достигать миллиардов долларов за одобренный препарат, [3] а полный процесс испытаний до одобрения может занять 7–15 лет. [4] [5] Спонсором может быть государственная организация или фармацевтическая , биотехнологическая или медицинская компания. Некоторые функции, необходимые для испытания, такие как мониторинг и лабораторные работы, могут управляться сторонним партнером, таким как контрактная исследовательская организация или центральная лаборатория. Только 10 процентов всех препаратов, запущенных в клинических испытаниях на людях, становятся одобренными препаратами . [6]
Некоторые клинические испытания включают здоровых субъектов без каких-либо ранее существовавших медицинских состояний . Другие клинические испытания относятся к людям с определенными заболеваниями, которые готовы попробовать экспериментальное лечение. Пилотные эксперименты проводятся для получения информации для дизайна последующего клинического испытания. [ необходима цитата ]
Существует две цели тестирования медицинских методов лечения: узнать, достаточно ли они эффективны, что называется «эффективностью» или «результативностью»; и узнать, достаточно ли они безопасны, что называется «безопасностью». [1] Ни один из этих критериев не является абсолютным; и безопасность, и эффективность оцениваются относительно того, как предполагается использовать лечение, какие другие методы лечения доступны и тяжесть заболевания или состояния. Преимущества должны перевешивать риски. [7] [8] : 8 Например, многие препараты для лечения рака имеют серьезные побочные эффекты, которые были бы неприемлемы для безрецептурных обезболивающих, однако препараты от рака были одобрены, поскольку они используются под наблюдением врача и используются при угрожающем жизни состоянии. [9]
В США пожилые люди составляют 14% населения, при этом они потребляют более трети лекарств. [10] Люди старше 55 лет (или аналогичного предельного возраста) часто исключаются из испытаний, поскольку их более серьезные проблемы со здоровьем и употребление наркотиков усложняют интерпретацию данных, а также потому, что у них другие физиологические возможности, чем у молодых людей. Дети и люди с не связанными с ними медицинскими состояниями также часто исключаются. [11] Беременные женщины часто исключаются из-за потенциальных рисков для плода .
Спонсор разрабатывает исследование совместно с группой экспертов-клиницистов-исследователей, включая альтернативные или существующие методы лечения для сравнения с новым препаратом и какие типы пациентов могут получить пользу. Если спонсор не может получить достаточное количество испытуемых в одном месте, исследователи в других местах набираются для участия в исследовании. [ необходима цитата ]
В ходе испытания исследователи набирают субъектов с заранее определенными характеристиками, назначают лечение(я) и собирают данные о здоровье субъектов в течение определенного периода времени. Данные включают такие измерения, как показатели жизнедеятельности , концентрацию исследуемого препарата в крови или тканях, изменения симптомов и происходит ли улучшение или ухудшение состояния, на которое нацелен исследуемый препарат. Исследователи отправляют данные спонсору испытания, который затем анализирует объединенные данные с помощью статистических тестов . [ необходима цитата ]
Примерами целей клинических испытаний являются оценка безопасности и относительной эффективности лекарственного средства или устройства: [ необходима ссылка ]
В то время как большинство клинических испытаний тестируют одну альтернативу новому вмешательству, некоторые расширяют их до трех или четырех и могут включать плацебо . [ необходима ссылка ]
За исключением небольших испытаний в одном месте, дизайн и цели указаны в документе, который называется протоколом клинического испытания . Протокол является «руководством по эксплуатации» испытания и гарантирует, что все исследователи проводят испытание одинаково на схожих субъектах и что данные сопоставимы по всем субъектам. [ необходима цитата ]
Поскольку испытание предназначено для проверки гипотез и строгого мониторинга и оценки результатов, его можно рассматривать как применение научного метода , в частности экспериментального этапа. [ необходима цитата ]
Наиболее распространенные клинические испытания оценивают новые фармацевтические продукты, медицинские устройства, биологические препараты , диагностические анализы , психотерапию или другие вмешательства. [12] Клинические испытания могут потребоваться до того, как национальный регулирующий орган [13] одобрит маркетинг инновации.
Подобно лекарствам, производители медицинских устройств в Соединенных Штатах обязаны проводить клинические испытания для предпродажного одобрения . [14] Испытания устройств могут сравнивать новое устройство с устоявшейся терапией или могут сравнивать похожие устройства друг с другом. Примером первого в области сосудистой хирургии является исследование Open versus Endovascular Repair (OVER) для лечения аневризмы брюшной аорты , в котором сравнивали старую технику открытого восстановления аорты с новым устройством для эндоваскулярного восстановления аневризмы . [15] Примером последнего являются клинические испытания механических устройств, используемых для лечения недержания мочи у взрослых женщин . [16]
Подобно лекарствам, медицинские или хирургические процедуры могут быть подвергнуты клиническим испытаниям, [17] например, сравнение различных хирургических подходов к лечению фибромиом при субфертильности . [18] Однако, когда клинические испытания неэтичны или логистически невозможны в хирургических условиях, исследования случай-контроль будут заменены. [19]
Помимо участия в клиническом испытании, представители общественности могут активно сотрудничать с исследователями в разработке и проведении клинических исследований . Это известно как участие пациентов и общественности (PPI). Участие общественности подразумевает рабочее партнерство между пациентами, лицами, осуществляющими уход, людьми с жизненным опытом, и исследователями для формирования и влияния на то, что и как исследуется. [20] PPI может улучшить качество исследований и сделать их более актуальными и доступными. Люди с текущим или прошлым опытом болезни могут предоставить другую точку зрения, чем профессионалы, и дополнить их знания. Благодаря своим личным знаниям они могут определить темы исследований, которые актуальны и важны для тех, кто живет с болезнью или пользуется услугой. Они также могут помочь сделать исследование более обоснованным в потребностях конкретных сообществ, частью которых они являются. Публичные участники также могут гарантировать, что исследование представлено на простом языке , понятном для более широкого общества и конкретных групп, для которых оно наиболее актуально. [21]

Хотя ранние медицинские эксперименты проводились часто, использование контрольной группы для обеспечения точного сравнения для демонстрации эффективности вмешательства, как правило, отсутствовало. Например, леди Мэри Уортли Монтегю , которая боролась за введение прививки (тогда называвшейся вариоляцией) для предотвращения оспы , организовала для семи заключенных, приговоренных к смертной казни, вариоляцию в обмен на их жизнь. Хотя они выжили и не заразились оспой, не было контрольной группы, чтобы оценить, был ли этот результат вызван прививкой или каким-то другим фактором. Аналогичные эксперименты, проведенные Эдвардом Дженнером над его вакциной от оспы, были столь же концептуально ошибочными. [22]
Первое надлежащее клиническое испытание было проведено шотландским врачом Джеймсом Линдом . [23] Болезнь цинга , которая, как теперь известно, вызывается дефицитом витамина С , часто имела ужасные последствия для благополучия экипажа дальних океанских путешествий. В 1740 году катастрофический результат кругосветного плавания Энсона привлек большое внимание в Европе; из 1900 человек 1400 умерли, большинство из них предположительно от заражения цингой. [24] Джон Вудол , английский военный хирург Британской Ост-Индской компании , рекомендовал употребление цитрусовых с 17 века, но их использование не стало широко распространенным. [25]
Линд провел первое систематическое клиническое испытание в 1747 году. [26] Он включил в эксперимент диетическую добавку кислотного качества после двух месяцев в море, когда корабль уже был поражен цингой. Он разделил двенадцать моряков, больных цингой, на шесть групп по два человека. Все они получали одинаковую диету, но, кроме того, группа один получала кварту сидра ежедневно , группа два — двадцать пять капель эликсира купороса ( серной кислоты ), группа три — шесть ложек уксуса , группа четыре — полпинты морской воды, группа пять получала два апельсина и один лимон , а последняя группа — пряную пасту и напиток из ячменной воды . Лечение группы пять прекратилось через шесть дней, когда у них закончились фрукты, но к тому времени один моряк был пригоден для службы, а другой почти выздоровел. Кроме того, только группа один также показала некоторый эффект от своего лечения. [27] Каждый год 20 мая отмечается как День клинических испытаний в честь исследований Линда. [28]
После 1750 года дисциплина начала приобретать свою современную форму. [29] [30] Английский врач Джон Хейгарт продемонстрировал важность контрольной группы для правильной идентификации эффекта плацебо в своем знаменитом исследовании неэффективного средства, называемого тракторами Перкина . Дальнейшая работа в этом направлении была проведена выдающимся врачом сэром Уильямом Галлом, 1-м баронетом в 1860-х годах. [22]
Фредерик Акбар Магомед (ум. 1884), работавший в больнице Гая в Лондоне , внес значительный вклад в процесс клинических испытаний, где «он отделил хронический нефрит с вторичной гипертензией от того, что мы сейчас называем эссенциальной гипертензией . Он также основал Коллективный отчет об исследованиях для Британской медицинской ассоциации ; эта организация собирала данные от врачей, практикующих за пределами больничных учреждений, и была предшественником современных совместных клинических испытаний». [31]

Идеи сэра Рональда А. Фишера до сих пор играют роль в клинических испытаниях. Работая на экспериментальной станции Ротамстеда в области сельского хозяйства, Фишер разработал свои Принципы экспериментального дизайна в 1920-х годах как точную методологию для надлежащего дизайна экспериментов. Среди его основных идей — важность рандомизации — случайного распределения людей по разным группам для эксперимента; [32] репликации — для уменьшения неопределенности измерения следует повторять, а эксперименты воспроизводить для выявления источников вариации; [33] блокирования — для объединения экспериментальных единиц в группы единиц, которые похожи друг на друга, и, таким образом, для сокращения нерелевантных источников вариации; использования факторных экспериментов — эффективно для оценки эффектов и возможных взаимодействий нескольких независимых факторов. [22] Из них блокирование и факторный дизайн редко применяются в клинических испытаниях, потому что экспериментальные единицы являются людьми и, как правило, существует только одно независимое вмешательство: лечение. [ требуется ссылка ]
Британский медицинский исследовательский совет официально признал важность клинических испытаний с 1930-х годов. Совет создал Комитет по терапевтическим испытаниям для консультирования и оказания помощи в организации должным образом контролируемых клинических испытаний новых продуктов, которые, по экспериментальным данным, могут иметь ценность в лечении заболеваний. [22]
Первое рандомизированное лечебное испытание было проведено в MRC Tuberculosis Research Unit сэром Джеффри Маршаллом (1887–1982). Испытание, проведенное между 1946 и 1947 годами, было направлено на проверку эффективности химического стрептомицина для лечения туберкулеза легких . Испытание было как двойным слепым , так и плацебо-контролируемым . [34]
Методология клинических испытаний была далее развита сэром Остином Брэдфордом Хиллом , который принимал участие в испытаниях стрептомицина. С 1920-х годов Хилл применял статистику в медицине, посещая лекции известного математика Карла Пирсона и других. Он прославился благодаря эпохальному исследованию, проведенному совместно с Ричардом Доллом по корреляции между курением и раком легких . Они провели исследование случай-контроль в 1950 году, в котором сравнивали пациентов с раком легких с соответствующим контролем, а также начали устойчивое долгосрочное перспективное исследование более широкой проблемы курения и здоровья, которое включало изучение привычек курения и здоровья более 30 000 врачей в течение нескольких лет. Его сертификат на избрание в Королевское общество называл его «... лидером в разработке в медицине точных экспериментальных методов, которые теперь используются на национальном и международном уровнях при оценке новых терапевтических и профилактических средств ».
Международный день клинических испытаний отмечается 20 мая. [35]
Аббревиатуры, используемые в названиях клинических испытаний , часто являются надуманными и стали предметом насмешек. [36]
Клинические испытания классифицируются по исследовательской цели, поставленной исследователями. [12]
Испытания классифицируются по их цели. После того, как одобрение на исследование на людях предоставлено спонсору испытания, Управление по контролю за продуктами и лекарствами США (FDA) организует и контролирует результаты испытаний в соответствии с типом: [12]
Клинические испытания обычно проводятся в четыре фазы, причем на каждой фазе задействовано разное количество субъектов, и каждая фаза имеет свою цель, направленную на выявление конкретного эффекта. [12]
Клинические испытания новых препаратов обычно делятся на пять фаз. Каждая фаза процесса одобрения препарата рассматривается как отдельное клиническое испытание. Процесс разработки препарата обычно проходит через фазы I–IV в течение многих лет, часто в течение десятилетия или дольше. Если препарат успешно проходит фазы I, II и III, он обычно одобряется национальным регулирующим органом для использования среди населения в целом. [12] Испытания фазы IV проводятся после того, как недавно одобренный препарат, диагностическое средство или устройство поступают на рынок, обеспечивая оценку рисков, преимуществ или наилучшего использования. [12]
Фундаментальное различие в практике, основанной на доказательствах, существует между наблюдательными исследованиями и рандомизированными контролируемыми испытаниями . [45] Типы наблюдательных исследований в эпидемиологии , такие как когортное исследование и исследование случай-контроль , предоставляют менее убедительные доказательства, чем рандомизированное контролируемое испытание. [45] В наблюдательных исследованиях исследователи ретроспективно оценивают связи между лечением, назначенным участникам, и их состоянием здоровья, что может привести к значительным ошибкам в дизайне и интерпретации. [46]
Рандомизированное контролируемое исследование может предоставить убедительные доказательства того, что исследуемое лечение оказывает влияние на здоровье человека. [45]
Некоторые испытания лекарственных препаратов фазы II и большинство испытаний лекарственных препаратов фазы III проводятся как рандомизированные, двойные слепые и плацебо- контролируемые. [ необходима ссылка ]
Клинические исследования с небольшим количеством субъектов могут «спонсироваться» отдельными исследователями или небольшой группой исследователей и предназначены для проверки простых вопросов или возможности расширения исследования для более полного рандомизированного контролируемого испытания. [47]
Клинические исследования могут быть «спонсированы» (финансированы и организованы) академическими институтами, фармацевтическими компаниями, государственными структурами и даже частными группами. Испытания проводятся для новых лекарств, биотехнологий, диагностических анализов или медицинских устройств, чтобы определить их безопасность и эффективность до представления на рассмотрение регулирующих органов, которое определит одобрение рынка. [ необходима цитата ]
В случаях, когда давать плацебо человеку, страдающему от заболевания, может быть неэтично, вместо этого могут быть проведены испытания с «активным компаратором» (также известные как «активный контроль»). [48] В испытаниях с активной контрольной группой субъектам дают либо экспериментальное лечение, либо ранее одобренное лечение с известной эффективностью. В других случаях спонсоры могут провести испытание с активным компаратором, чтобы установить эффективность, заявленную относительно активного компаратора вместо плацебо в маркировке . [ необходима цитата ]
Основной протокол включает несколько подисследований, которые могут иметь разные цели и включать скоординированные усилия по оценке одного или нескольких медицинских продуктов при одном или нескольких заболеваниях или состояниях в рамках общей структуры исследования. Испытания, которые могли бы разработать основной протокол, включают зонтичное испытание (несколько медицинских продуктов для одного заболевания), платформенное испытание (несколько продуктов для одного заболевания, входящих и выходящих из платформы) и корзиночное испытание (один медицинский продукт для нескольких заболеваний или подтипов заболеваний). [49]
Генетическое тестирование позволяет исследователям группировать пациентов в соответствии с их генетическим профилем, доставлять препараты на основе этого профиля этой группе и сравнивать результаты. Участвовать могут несколько компаний, каждая из которых представляет свой препарат. Первый такой подход нацелен на плоскоклеточный рак , который включает в себя различные генетические нарушения от пациента к пациенту. Участвуют Amgen, AstraZeneca и Pfizer, впервые они работали вместе в поздней стадии исследования. Пациенты, чьи геномные профили не соответствуют ни одному из исследуемых препаратов, получают препарат, предназначенный для стимуляции иммунной системы для атаки на рак. [50]
Протокол клинического испытания — это документ, используемый для определения и управления испытанием. Он готовится группой экспертов. Все исследователи исследования должны строго соблюдать протокол. [ необходима цитата ]
Протокол описывает научное обоснование, цель(и), дизайн, методологию, статистические соображения и организацию запланированного испытания. Подробности испытания приведены в документах, на которые даны ссылки в протоколе, например, в брошюре исследователя . [ необходима цитата ]
Протокол содержит точный план исследования, чтобы гарантировать безопасность и здоровье испытуемых и предоставить точный шаблон для проведения исследования исследователями. Это позволяет объединить данные по всем исследователям/сайтам. Протокол также информирует администраторов исследования (часто контрактную исследовательскую организацию ). [ необходима цитата ]
Формат и содержание протоколов клинических испытаний, спонсируемых фармацевтическими, биотехнологическими или медицинскими компаниями в США, Европейском Союзе или Японии, были стандартизированы в соответствии с руководством по надлежащей клинической практике [51], выпущенным Международной конференцией по гармонизации (ICH). [52] Регулирующие органы в Канаде , Китае , Южной Корее и Великобритании также следуют рекомендациям ICH. Такие журналы, как Trials , поощряют исследователей публиковать свои протоколы.

Клинические испытания набирают субъектов исследования для подписания документа, представляющего их « информированное согласие ». [53] Документ включает в себя такие детали, как его цель, продолжительность, требуемые процедуры, риски, потенциальные преимущества, ключевые контакты и институциональные требования. [54] Затем участник решает, подписывать ли документ. Документ не является контрактом, так как участник может отказаться от участия в любое время без штрафных санкций. [ необходима цитата ]
Информированное согласие — это юридический процесс, в котором рекрута инструктируют о ключевых фактах, прежде чем он примет решение об участии. [53] Исследователи объясняют детали исследования в терминах, которые субъект может понять. Информация представлена на родном языке субъекта. Как правило, дети не могут самостоятельно предоставить информированное согласие, но в зависимости от их возраста и других факторов от них может потребоваться предоставить информированное согласие. [ необходима цитата ]
В любом клиническом исследовании количество субъектов, также называемое размером выборки, оказывает большое влияние на способность надежно обнаруживать и измерять эффекты вмешательства. Эта способность описывается как его « мощность », которая должна быть рассчитана до начала исследования, чтобы выяснить, стоит ли исследование своих затрат. [55] В целом, больший размер выборки увеличивает статистическую мощность, а также стоимость.
Статистическая мощность оценивает способность испытания обнаруживать разницу определенного размера (или больше) между группами лечения и контроля. Например, испытание препарата, снижающего уровень липидов , против плацебо со 100 пациентами в каждой группе может иметь мощность 0,90 для обнаружения разницы между плацебо и группами испытания, получающими дозировку 10 мг/дл или более, но только 0,70 для обнаружения разницы в 6 мг/дл. [ необходима цитата ]
Простое предоставление лечения может иметь неспецифические эффекты. Они контролируются включением пациентов, которые получают только плацебо. Субъекты распределяются случайным образом, без информирования их о том, к какой группе они принадлежат. Многие испытания проводятся двойным слепым методом, так что исследователи не знают, к какой группе отнесен субъект.
Назначение субъекта в группу плацебо может представлять этическую проблему, если это нарушает его или ее право на получение наилучшего доступного лечения. Хельсинкская декларация содержит руководящие принципы по этому вопросу.
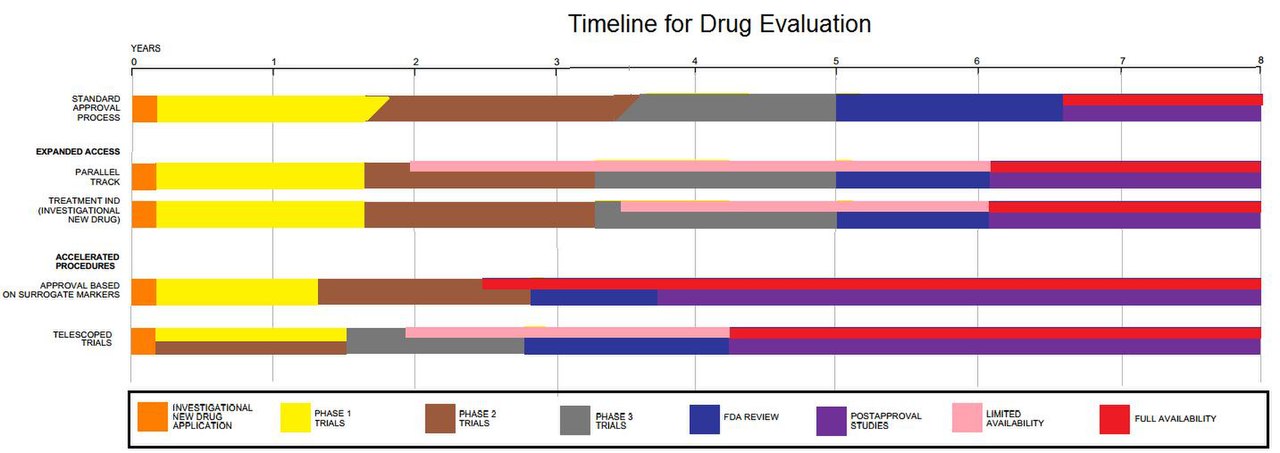
Клинические испытания — это лишь малая часть исследований, которые идут на разработку нового лечения. Например, потенциальные препараты сначала должны быть обнаружены, очищены, охарактеризованы и протестированы в лабораториях (в исследованиях на клетках и животных) до того, как они когда-либо пройдут клинические испытания. В целом, около 1000 потенциальных препаратов тестируются, прежде чем хотя бы один из них достигнет точки тестирования в клинических испытаниях. [56] Например, новое лекарство от рака в среднем имеет шесть лет исследований за собой, прежде чем оно даже попадает на клинические испытания. Но основная задержка в обеспечении доступности новых лекарств от рака — это время, необходимое для завершения самих клинических испытаний. В среднем проходит около восьми лет с момента, когда лекарство от рака попадает на клинические испытания, до того, как оно получает одобрение регулирующих органов на продажу населению. [57] Лекарства от других заболеваний имеют схожие сроки.
Некоторые причины, по которым клиническое исследование может длиться несколько лет:
Клиническое исследование может также включать расширенный период наблюдения после исследования, длящийся от нескольких месяцев до нескольких лет, для людей, которые участвовали в исследовании, так называемую «фазу расширения», целью которой является определение долгосрочного воздействия лечения. [58]
Самым большим препятствием для завершения исследований является нехватка людей, которые принимают участие. Все испытания лекарств и многих устройств нацелены на подгруппу населения, что означает, что не все могут участвовать. Некоторые испытания лекарств требуют, чтобы у пациентов были необычные комбинации характеристик заболевания. Сложно найти подходящих пациентов и получить их согласие, особенно когда они могут не получить прямой выгоды (потому что им не платят, исследуемый препарат еще не доказал свою эффективность или пациент может получать плацебо). В случае онкологических больных менее 5% взрослых с раком будут участвовать в испытаниях лекарств. По данным Pharmaceutical Research and Manufacturers of America (PhRMA), около 400 лекарств от рака проходили клинические испытания в 2005 году. Не все из них окажутся полезными, но те, которые окажутся полезными, могут быть отложены в получении одобрения из-за слишком малого числа участников. [59]
Для клинических испытаний, включающих потенциальные сезонные влияния (такие как воздушная аллергия , сезонное аффективное расстройство , грипп и кожные заболевания ), исследование может проводиться в течение ограниченного периода года (например, весной для пыльцевой аллергии), когда можно протестировать препарат. [60] [61]
Клинические испытания, в которых не используются новые лекарственные препараты, обычно длятся гораздо меньше. (Исключением являются эпидемиологические исследования, такие как Исследование здоровья медсестер ).
Клинические испытания, разработанные местным исследователем, и (в США) клинические испытания, финансируемые из федерального бюджета, почти всегда администрируются исследователем, который разработал исследование и подал заявку на грант. Исследования с использованием небольших устройств могут администрироваться спонсирующей компанией. Клинические испытания новых препаратов обычно администрируются контрактной исследовательской организацией (CRO), нанятой спонсирующей компанией. Спонсор обеспечивает надзор за лекарственными средствами и медицинским обслуживанием. CRO нанимается на выполнение всей административной работы по клиническому испытанию. Для фаз II–IV CRO набирает участвующих исследователей, обучает их, предоставляет им расходные материалы, координирует администрирование исследования и сбор данных , организует встречи, контролирует соответствие центров клиническим протоколам и обеспечивает получение спонсором данных из каждого центра. Специализированные организации по управлению центрами также могут быть наняты для координации с CRO с целью обеспечения быстрого одобрения IRB/IEC и более быстрого инициирования центра и набора пациентов. Клинические испытания фазы I новых лекарств часто проводятся в специализированной клинике клинических испытаний с привлеченными фармакологами, где субъекты могут наблюдаться штатным персоналом. Эти клиники часто управляются CRO, которая специализируется на этих исследованиях.
На участвующем сайте один или несколько научных сотрудников (часто медсестер) выполняют большую часть работы по проведению клинического исследования. Работа научного сотрудника может включать некоторые или все из следующих действий: предоставление местному институциональному наблюдательному совету (IRB) документации, необходимой для получения разрешения на проведение исследования, помощь в запуске исследования, выявление подходящих пациентов, получение согласия от них или их семей, проведение исследуемого лечения(й), сбор и статистический анализ данных, ведение и обновление файлов данных во время последующего наблюдения и общение с IRB, а также со спонсором и CRO.
В контексте клинического испытания качество обычно относится к отсутствию ошибок, которые могут повлиять на принятие решений, как во время проведения испытания, так и при использовании результатов испытания. [62]
Модель интерактивной справедливости может использоваться для проверки эффектов готовности говорить с врачом о зачислении в клинические испытания. [63] Результаты показали, что потенциальные кандидаты на клинические испытания с меньшей вероятностью будут зачислены в клинические испытания, если пациент более готов говорить со своим врачом. Причиной этого открытия может быть то, что пациенты довольны своим текущим лечением. Еще одной причиной отрицательной связи между воспринимаемой справедливостью и зачислением в клинические испытания является отсутствие независимости от поставщика услуг. Результаты показали, что существует положительная связь между отсутствием готовности говорить со своим врачом и зачислением в клинические испытания. Отсутствие готовности говорить о клинических испытаниях с текущими поставщиками услуг может быть связано с независимостью пациентов от врача. Пациенты, которые с меньшей вероятностью говорят о клинических испытаниях, с большей готовностью используют другие источники информации, чтобы получить лучшее представление об альтернативных методах лечения. Зачисление в клинические испытания должно быть мотивировано использованием веб-сайтов и телевизионной рекламы для информирования общественности о зачислении в клинические испытания.
В последнее десятилетие наблюдается распространение использования информационных технологий в планировании и проведении клинических испытаний. Системы управления клиническими испытаниями часто используются спонсорами исследований или CRO для помощи в планировании и управлении операционными аспектами клинического испытания, особенно в отношении исследовательских центров. Расширенная аналитика для определения исследователей и исследовательских центров с опытом в определенной области использует общедоступную и частную информацию о текущих исследованиях. [64] Веб- системы электронного сбора данных (EDC) и управления клиническими данными используются в большинстве клинических испытаний [65] для сбора данных отчетов о случаях с сайтов, управления их качеством и подготовки к анализу. Интерактивные системы голосового ответа используются сайтами для регистрации набора пациентов с помощью телефона и распределения пациентов по определенным группам лечения (хотя телефоны все чаще заменяются веб-инструментами (IWRS), которые иногда являются частью системы EDC). В то время как результаты, сообщаемые пациентами, в прошлом часто были на бумажных носителях, измерения все чаще собираются с помощью веб-порталов или портативных устройств ePRO (или eDiary), иногда беспроводных. [66] Статистическое программное обеспечение используется для анализа собранных данных и подготовки их к подаче в регулирующие органы. Доступ ко многим из этих приложений все чаще агрегируется на веб- порталах клинических испытаний . В 2011 году FDA одобрило испытание фазы I, в котором использовался телемониторинг, также известный как удаленный мониторинг пациентов, для сбора биометрических данных в домах пациентов и их передачи в электронном виде в базу данных испытаний. Эта технология обеспечивает гораздо больше точек данных и гораздо удобнее для пациентов, поскольку им приходится реже посещать места испытаний. Как отмечено ниже, децентрализованные клинические испытания — это те, которые не требуют физического присутствия пациентов на месте, а вместо этого в значительной степени полагаются на сбор цифровых данных о состоянии здоровья, процессы цифрового информированного согласия и т. д.
Клиническое исследование дает данные, которые могут выявить количественные различия между двумя или более вмешательствами; статистический анализ используется для определения того, являются ли такие различия истинными, случайными или такими же, как отсутствие лечения (плацебо). [67] [68] Данные клинического исследования накапливаются постепенно в течение всего исследования, от нескольких месяцев до нескольких лет. [53] Соответственно, результаты для участников, набранных на ранних этапах исследования, становятся доступными для анализа, пока субъекты все еще распределяются по группам лечения в исследовании. Ранний анализ может позволить появляющимся доказательствам помочь в принятии решений о том, следует ли прекратить исследование или переназначить участников в более успешный сегмент исследования. [67] Исследователи также могут захотеть остановить исследование, если анализ данных не показывает никакого эффекта от лечения. [68]
Клинические испытания находятся под пристальным наблюдением соответствующих регулирующих органов. Все исследования, включающие медицинское или терапевтическое вмешательство в работу пациентов, должны быть одобрены контролирующим этическим комитетом до выдачи разрешения на проведение испытания. Местный этический комитет имеет право самостоятельно контролировать неинтервенционные исследования (наблюдательные исследования или исследования, использующие уже собранные данные). В США этот орган называется Институциональным наблюдательным советом (IRB); в ЕС они называются этическими комитетами . Большинство IRB находятся в местной больнице или учреждении исследователя, но некоторые спонсоры разрешают использовать центральный (независимый/коммерческий) IRB для исследователей, работающих в небольших учреждениях.
Чтобы быть этичными, исследователи должны получить полное и информированное согласие участвующих людей-субъектов. (Одной из основных функций IRB является обеспечение адекватной информированности потенциальных пациентов о клиническом исследовании.) Если пациент не может дать согласие сам, исследователи могут запросить согласие у законного представителя пациента. Кроме того, участники клинического исследования должны быть осведомлены о том, что они могут отказаться от клинического исследования в любое время без каких-либо неблагоприятных действий против них. [69] В Калифорнии штат отдал приоритет лицам, которые могут выступать в качестве законного представителя. [70]
В некоторых регионах США местные IRB должны сертифицировать исследователей и их персонал, прежде чем они смогут проводить клинические испытания. Они должны понимать федеральный закон о конфиденциальности пациентов ( HIPAA ) и надлежащую клиническую практику. Руководящие принципы Международной конференции по гармонизации надлежащей клинической практики представляют собой набор стандартов, используемых на международном уровне для проведения клинических испытаний. Руководящие принципы направлены на обеспечение «защиты прав, безопасности и благополучия субъектов испытаний».
Понятие информированного согласия на участие в исследовании людей существует во многих странах, но его точное определение может различаться.
Информированное согласие, несомненно, является «необходимым» условием этичного поведения, но не «гарантирует» этичное поведение. В испытаниях по сострадательному использованию последнее становится особенно сложной проблемой. Конечная цель — служить сообществу пациентов или будущих пациентов наилучшим и наиболее ответственным образом. См. также Расширенный доступ . Однако может быть сложно превратить эту цель в четко определенную, количественную, целевую функцию. Однако в некоторых случаях это можно сделать, например, для вопросов о том, когда следует прекратить последовательное лечение (см. Алгоритм шансов ), и тогда количественные методы могут сыграть важную роль.
Дополнительные этические проблемы возникают при проведении клинических испытаний на детях ( педиатрия ), а также в чрезвычайных или эпидемических ситуациях. [71] [72]
Этическое уравновешивание прав нескольких заинтересованных сторон может быть сложным. Например, когда испытания лекарств терпят неудачу, спонсоры могут быть обязаны немедленно сообщить об этом текущим и потенциальным инвесторам, что означает, что и исследовательский персонал, и зарегистрированные участники могут первыми услышать об окончании испытания из новостей публичного бизнеса . [73]
В ответ на конкретные случаи, когда неблагоприятные данные исследований, спонсируемых фармацевтическими компаниями, не были опубликованы, Pharmaceutical Research and Manufacturers of America опубликовала новые руководящие принципы, призывающие компании сообщать обо всех результатах и ограничивать финансовое участие исследователей в фармацевтических компаниях. [74] Конгресс США подписал законопроект, который требует, чтобы клинические испытания фазы II и фазы III были зарегистрированы спонсором на веб-сайте clinicaltrials.gov, составленном Национальными институтами здравоохранения . [75]
Исследователи лекарственных препаратов, не работающие напрямую в фармацевтических компаниях, часто ищут гранты у производителей, а производители часто обращаются к академическим исследователям для проведения исследований в сетях университетов и их больниц, например, для трансляционных исследований рака. Аналогичным образом, конкуренция за постоянные академические должности, государственные гранты и престиж создают конфликты интересов среди академических ученых. [76] Согласно одному исследованию, примерно 75% статей, отозванных по причинам, связанным с неправомерным поведением, не имеют заявленной финансовой поддержки со стороны отрасли. [77] Испытания посева особенно противоречивы. [78]
В Соединенных Штатах все клинические испытания, представленные в FDA в рамках процесса одобрения лекарственных препаратов, проходят независимую оценку клинических экспертов Управления по контролю за продуктами питания и лекарственными средствами [79] , включая проверки первичного сбора данных в выбранных местах проведения клинических испытаний. [80]
В 2001 году редакторы 12 крупных журналов выпустили совместную редакционную статью, опубликованную в каждом журнале, о контроле над клиническими испытаниями, осуществляемом спонсорами, в частности, нацеленную на использование контрактов, которые позволяют спонсорам просматривать исследования до публикации и воздерживаться от публикации. Они усилили редакционные ограничения, чтобы противостоять эффекту. В редакционной статье отмечалось, что к 2000 году организации, занимающиеся контрактными исследованиями, получили 60% грантов от фармацевтических компаний в США. Исследователи могут быть ограничены в возможности вносить вклад в дизайн испытаний, получать доступ к необработанным данным и интерпретировать результаты. [81]
Несмотря на явные рекомендации заинтересованных сторон о мерах по улучшению стандартов медицинских исследований, спонсируемых промышленностью, [82] в 2013 году Тоэн предупредил о сохраняющемся пробеле в достоверности выводов, вытекающих из финансируемых промышленностью клинических испытаний, и призвал к обеспечению строгого соблюдения этических стандартов в промышленном сотрудничестве с академическими кругами, чтобы избежать дальнейшего подрыва доверия общественности. [83] Вопросы, на которые следует обратить внимание в этом отношении, включают потенциальную ошибку наблюдения, продолжительность времени наблюдения для поддерживающих исследований, выбор групп пациентов, факторы, которые влияют на реакцию плацебо, и источники финансирования. [84] [85] [86]
Проведение клинических испытаний вакцин во время эпидемий и пандемий является предметом этических проблем. Для болезней с высоким уровнем смертности, таких как Эбола, назначение людей в плацебо или контрольную группу может рассматриваться как смертный приговор. В ответ на этические проблемы, связанные с клиническими исследованиями во время эпидемий, Национальная медицинская академия подготовила отчет, в котором определены семь этических и научных соображений. Эти соображения следующие: [87]
Беременные женщины и дети обычно исключаются из клинических испытаний как уязвимые группы населения, хотя данные, подтверждающие их исключение, не являются надежными. Исключая их из клинических испытаний, информация о безопасности и эффективности терапии для этих групп населения часто отсутствует. В ранней истории эпидемии ВИЧ/СПИДа ученый отметил, что, исключая эти группы из потенциально спасающего жизнь лечения, они были «защищены до смерти». Такие проекты, как Research Ethics for Vaccines, Epidemics, and New Technologies (PREVENT), выступали за этическое включение беременных женщин в испытания вакцин. Включение детей в клинические испытания имеет дополнительные моральные соображения, поскольку дети не имеют самостоятельности в принятии решений. Испытания в прошлом подвергались критике за использование госпитализированных детей или сирот; эти этические проблемы фактически остановили будущие исследования. В целях поддержания эффективной педиатрической помощи несколько европейских стран и США проводят политику, побуждающую или заставляющую фармацевтические компании проводить педиатрические испытания. Международное руководство рекомендует проводить этические педиатрические испытания, ограничивая вред, учитывая различные риски и принимая во внимание сложность педиатрической помощи. [87]
Ответственность за безопасность субъектов клинического испытания делится между спонсором, местными исследователями (если они не являются спонсором), различными ЭСО, которые контролируют исследование, и (в некоторых случаях, если исследование касается продаваемого препарата или устройства) регулирующим органом страны, где препарат или устройство будут продаваться.
Систематический параллельный обзор безопасности часто используется для обеспечения безопасности участников исследования. Проведение и текущий обзор разрабатываются так, чтобы быть пропорциональными риску исследования. Обычно эта роль выполняется Комитетом по данным и безопасности , назначенным извне медицинским монитором безопасности, [88] Независимым сотрудником по безопасности или для небольших или низкорисковых исследований главным исследователем. [89]
В целях безопасности многие клинические испытания лекарств [90] разработаны так, чтобы исключить женщин детородного возраста, беременных женщин или женщин, которые забеременели во время исследования. В некоторых случаях партнеры-мужчины этих женщин также исключаются или должны принимать меры по контролю рождаемости.
На протяжении всего клинического испытания спонсор несет ответственность за точное информирование местных исследователей об истинных исторических данных о безопасности препарата, устройства или других медицинских методов лечения, которые будут тестироваться, и о любых потенциальных взаимодействиях исследуемого лечения с уже одобренными методами лечения. Это позволяет местным исследователям принимать обоснованное решение о том, участвовать в исследовании или нет. Спонсор также несет ответственность за мониторинг результатов исследования по мере их поступления из различных центров по мере проведения испытания. В более крупных клинических испытаниях спонсор будет пользоваться услугами комитета по мониторингу данных (DMC, известного в США как совет по мониторингу безопасности данных). Эта независимая группа врачей и статистиков периодически встречается для обзора неослепленных данных, полученных спонсором к настоящему моменту. DMC имеет право рекомендовать прекращение исследования на основе своего обзора, например, если исследуемое лечение приводит к большему количеству смертей, чем стандартное лечение, или, по-видимому, вызывает непредвиденные и связанные с исследованием серьезные нежелательные явления . Спонсор несет ответственность за сбор отчетов о нежелательных явлениях от всех исследователей, участвующих в исследовании, и за информирование всех исследователей о своем мнении относительно того, связаны ли эти нежелательные явления с исследуемым лечением или нет.
Спонсор и местные исследователи несут совместную ответственность за написание информированного согласия , специфичного для данного места , которое точно информирует потенциальных субъектов об истинных рисках и потенциальных преимуществах участия в исследовании, в то же время представляя материал как можно более кратко и на обычном языке. Правила FDA гласят, что участие в клинических испытаниях является добровольным, при этом субъект имеет право не участвовать или прекратить участие в любое время. [91]
Этический принцип primum non-nocere («прежде всего, не навреди») направляет исследование, и если исследователь считает, что исследуемое лечение может нанести вред субъектам исследования, он может прекратить участие в любое время. С другой стороны, исследователи часто имеют финансовую заинтересованность в наборе субъектов и могут действовать неэтично, чтобы получить и сохранить их участие.
Местные исследователи несут ответственность за проведение исследования в соответствии с протоколом исследования и контроль за персоналом исследования на протяжении всего исследования. Местный исследователь или его/ее персонал исследования также несут ответственность за то, чтобы потенциальные субъекты исследования понимали риски и потенциальные преимущества участия в исследовании. Другими словами, они (или их законные представители) должны дать действительно информированное согласие.
Местные исследователи несут ответственность за рассмотрение всех отчетов о нежелательных явлениях, отправленных спонсором. Эти отчеты о нежелательных явлениях содержат мнения как исследователя (в месте, где произошло нежелательное явление), так и спонсора относительно связи нежелательного явления с исследуемым лечением. Местные исследователи также несут ответственность за вынесение независимого суждения по этим отчетам и за незамедлительное информирование местного IRB обо всех серьезных и связанных с исследуемым лечением нежелательных явлениях.
Когда спонсором является местный исследователь, формальных отчетов о нежелательных явлениях может не быть, но сотрудники исследования во всех местах несут ответственность за информирование координирующего исследователя о любых неожиданных событиях. Местный исследователь несет ответственность за то, чтобы быть правдивым перед местным IRB во всех сообщениях, касающихся исследования.
Одобрение Институционального наблюдательного совета (IRB) или Независимого этического комитета (IEC) необходимо до начала всех исследований, кроме самых неформальных. В коммерческих клинических испытаниях протокол исследования не утверждается IRB до того, как спонсор наберет центры для проведения испытания. Однако протокол исследования и процедуры были адаптированы для соответствия общим требованиям подачи IRB. В этом случае, а также при отсутствии независимого спонсора, каждый местный исследователь сайта представляет протокол исследования, согласие(я), формы сбора данных и подтверждающую документацию в местный IRB. Университеты и большинство больниц имеют внутренние IRB. Другие исследователи (например, в клиниках без предварительной записи) используют независимых IRB.
IRB тщательно изучает исследование как с точки зрения медицинской безопасности, так и с точки зрения защиты пациентов, участвующих в исследовании, прежде чем разрешить исследователю начать исследование. Это может потребовать внесения изменений в процедуры исследования или в объяснения, данные пациенту. Требуемый ежегодный отчет «постоянного обзора» от исследователя информирует IRB о ходе исследования и любой новой информации по безопасности, связанной с исследованием.
В США FDA может проводить аудит файлов местных исследователей после того, как они закончили участие в исследовании, чтобы проверить, правильно ли они следовали процедурам исследования. Этот аудит может быть случайным или по причине (потому что исследователь подозревается в мошенничестве с данными). Избегание аудита является стимулом для исследователей следовать процедурам исследования. «Охваченное клиническое исследование» относится к испытанию, представленному в FDA как часть маркетинговой заявки (например, как часть NDA или 510 (k) ), в отношении которого FDA может потребовать раскрытия финансовой заинтересованности клинического исследователя в результатах исследования. Например, заявитель должен раскрыть, владеет ли исследователь капиталом в спонсоре или владеет ли имущественным интересом в исследуемом продукте. FDA определяет охватываемое исследование как «... любое исследование лекарственного препарата, биологического продукта или устройства на людях, представленное в маркетинговой заявке или ходатайстве о реклассификации, на которое заявитель или FDA опираются для установления эффективности продукта (включая исследования, которые показывают эквивалентность эффективному продукту) или любое исследование, в котором один исследователь вносит значительный вклад в демонстрацию безопасности». [92]
В качестве альтернативы многие американские фармацевтические компании перенесли некоторые клинические испытания за границу. Преимущества проведения испытаний за границей включают более низкие затраты (в некоторых странах) и возможность проводить более крупные испытания в более короткие сроки, в то время как потенциальный недостаток заключается в более низком качестве управления испытаниями. [93] В разных странах действуют разные нормативные требования и возможности обеспечения соблюдения. По оценкам, 40% всех клинических испытаний в настоящее время проводятся в Азии, Восточной Европе, Центральной и Южной Америке. «В этих странах нет обязательной системы регистрации клинических испытаний, и многие не следуют европейским директивам в своей деятельности», - говорит Якоб Сийтсма из WEMOS, правозащитной организации здравоохранения, отслеживающей клинические испытания в развивающихся странах, базирующейся в Нидерландах. [94]
Начиная с 1980-х годов, гармонизация протоколов клинических испытаний была показана как осуществимая во всех странах Европейского союза. В то же время координация между Европой, Японией и Соединенными Штатами привела к совместной инициативе регуляторов и промышленности по международной гармонизации, названной в 1990 году Международной конференцией по гармонизации технических требований к регистрации фармацевтических препаратов для использования человеком (ICH) [95] В настоящее время большинство программ клинических испытаний следуют рекомендациям ICH, направленным на «обеспечение того, чтобы качественные, безопасные и эффективные лекарства разрабатывались и регистрировались наиболее эффективным и экономически выгодным способом. Эти мероприятия проводятся в интересах потребителя и общественного здравоохранения, чтобы предотвратить ненужное дублирование клинических испытаний на людях и минимизировать использование испытаний на животных, не ставя под угрозу нормативные обязательства по безопасности и эффективности». [96]
Агрегирование данных по безопасности в ходе клинических испытаний во время разработки препарата важно, поскольку испытания обычно разрабатываются с целью определения того, насколько хорошо работает препарат. Данные по безопасности, собранные и агрегированные в ходе нескольких испытаний по мере разработки препарата, позволяют спонсору, исследователям и регулирующим органам отслеживать совокупный профиль безопасности экспериментальных лекарств по мере их разработки. Ценность оценки совокупных данных по безопасности заключается в следующем: а) решения, основанные на совокупной оценке безопасности во время разработки препарата, могут приниматься на протяжении всей разработки препарата и б) это хорошо настраивает спонсора и регулирующие органы на оценку безопасности препарата после его одобрения. [97] [98] [99] [100] [101]
Стоимость клинических испытаний варьируется в зависимости от фазы испытания, типа испытания и изучаемого заболевания. Исследование клинических испытаний, проведенное в Соединенных Штатах с 2004 по 2012 год, показало, что средняя стоимость испытаний фазы I составляет от 1,4 до 6,6 млн долларов США в зависимости от типа заболевания. Испытания фазы II стоят от 7 до 20 млн долларов США, а испытания фазы III — от 11 до 53 млн долларов США. [102]
Стоимость исследования зависит от многих факторов, особенно от количества центров, проводящих исследование, количества вовлеченных пациентов и того, одобрен ли уже исследуемый метод лечения для медицинского применения.
Расходы, понесенные фармацевтической компанией при проведении клинических испытаний фазы III или IV, могут включать, среди прочего:
Эти расходы производятся в течение нескольких лет.
В США спонсоры могут получить 50-процентный налоговый кредит на клинические испытания, проводимые с препаратами, разрабатываемыми для лечения орфанных заболеваний . [103] Национальные агентства здравоохранения, такие как Национальные институты здравоохранения США , предлагают гранты исследователям, которые разрабатывают клинические испытания, которые пытаются ответить на исследовательские вопросы, представляющие интерес для агентства. В этих случаях исследователь, который пишет грант и администрирует исследование, выступает в качестве спонсора и координирует сбор данных с любых других сайтов. Эти другие сайты могут получать или не получать оплату за участие в исследовании, в зависимости от суммы гранта и объема усилий, ожидаемых от них. Использование интернет-ресурсов может, в некоторых случаях, снизить экономическое бремя. [104]
Исследователи часто получают компенсацию за свою работу в клинических испытаниях. Эти суммы могут быть небольшими, покрывая лишь частичную зарплату ассистентов исследователей и стоимость любых расходных материалов (обычно в случае исследований национального агентства здравоохранения), или быть существенными и включать «накладные расходы», которые позволяют исследователю платить исследовательскому персоналу в периоды между клиническими испытаниями. [ необходима цитата ]
Участники испытаний лекарственных препаратов фазы I не получают никакой прямой пользы для здоровья от участия. Им, как правило, платят за их время, причем платежи регулируются и не связаны с каким-либо риском. Мотивация здоровых добровольцев не ограничивается финансовым вознаграждением и может включать другие мотивы, такие как вклад в науку и другие. [105] На более поздних фазах испытаний субъектам может не выплачиваться плата за обеспечение их мотивации к участию с потенциальной пользой для здоровья или вкладом в медицинские знания. Небольшие выплаты могут производиться за расходы, связанные с исследованием, такие как поездки, или в качестве компенсации за их время, потраченное на предоставление последующей информации о состоянии их здоровья после окончания лечения в рамках испытания.

Испытания лекарственных препаратов фазы 0 и фазы I ищут здоровых добровольцев. Большинство других клинических испытаний ищут пациентов, которые имеют определенное заболевание или медицинское состояние. Разнообразие, наблюдаемое в обществе, должно быть отражено в клинических испытаниях посредством соответствующего включения этнических меньшинств . [106] Набор пациентов или набор участников играет важную роль в деятельности и обязанностях мест проведения клинических испытаний. [107]
Все добровольцы, рассматриваемые для участия в исследовании, должны пройти медицинское обследование. Требования различаются в зависимости от потребностей исследования, но обычно добровольцы проходят обследование в медицинской лаборатории на: [108]
Было отмечено, что участники клинических испытаний непропорционально белые. [109] [110] Часто меньшинства не информируются о клинических испытаниях. [111] Один из недавних систематических обзоров литературы показал, что раса/этническая принадлежность, а также пол не были хорошо представлены, а иногда даже отслеживались в качестве участников большого количества клинических испытаний лечения потери слуха у взрослых. [112] Это может снизить достоверность результатов в отношении небелых пациентов [113], поскольку неадекватно представляют большую часть населения .
В зависимости от требуемого типа участников спонсоры клинических испытаний или контрактные исследовательские организации, работающие от их имени, пытаются найти сайты с квалифицированным персоналом, а также доступ к пациентам, которые могли бы принять участие в испытании. Работая с этими сайтами, они могут использовать различные стратегии набора, включая базы данных пациентов, газетные и радиорекламы, листовки, плакаты в местах, куда могут пойти пациенты (например, в кабинетах врачей), и личный набор пациентов исследователями.
Добровольцы с определенными состояниями или заболеваниями имеют дополнительные онлайн-ресурсы, которые помогают им находить клинические испытания. Например, Fox Trial Finder связывает испытания болезни Паркинсона по всему миру с добровольцами, имеющими определенный набор критериев, таких как местоположение, возраст и симптомы. [114] Существуют и другие услуги, связанные с заболеваниями, для добровольцев, чтобы найти испытания, связанные с их состоянием. [115] Добровольцы могут искать непосредственно на ClinicalTrials.gov, чтобы найти испытания, используя реестр, который ведут Национальные институты здравоохранения США и Национальная медицинская библиотека . Существует также программное обеспечение, которое позволяет врачам находить варианты испытаний для отдельного пациента на основе таких данных, как геномные данные. [116]

Модель поиска и обработки информации о рисках (RISP) анализирует социальные последствия, которые влияют на отношение и принятие решений, касающихся клинических испытаний. [117] Люди, которые имеют более высокую ставку или интерес к лечению, предоставляемому в ходе клинического испытания, показали большую вероятность поиска информации о клинических испытаниях. Пациенты с раком сообщили о более оптимистичном отношении к клиническим испытаниям, чем население в целом. Наличие более оптимистичного взгляда на клинические испытания также приводит к большей вероятности зачисления. [117]
Сопоставление включает в себя систематическое сравнение клинической и демографической информации пациента с критериями приемлемости различных испытаний. Методы включают:
Хотя испытания обычно проводятся в крупных медицинских центрах, некоторые участники исключаются из-за расстояния и расходов, необходимых для поездки, что приводит к трудностям, невыгодному положению и неравенству для участников, особенно в сельских и недостаточно обслуживаемых общинах. Поэтому концепция «децентрализованного клинического испытания», которая сводит к минимуму или устраняет необходимость для пациентов ездить на места, [122] в настоящее время более распространена, возможность улучшена благодаря телемедицине и носимым технологиям . [123]
всех препаратов, начавших клинические испытания на людях, только 10 процентов получили одобрение FDA.
...
{{cite journal}}: CS1 maint: DOI неактивен по состоянию на ноябрь 2024 г. ( ссылка )