Бета-талассемии ( β-талассемии ) — это группа наследственных заболеваний крови . Это формы талассемии, вызванные снижением или отсутствием синтеза бета - цепей гемоглобина , которые приводят к различным результатам: от тяжелой анемии до клинически бессимптомных лиц. Глобальная ежегодная заболеваемость оценивается в один случай на 100 000. [4] Бета-талассемии возникают из-за сбоев в работе субъединицы гемоглобина бета или HBB. Тяжесть заболевания зависит от характера мутации. [5]
Неспособность организма строить новые бета-цепи приводит к недопроизводству HbA (взрослого гемоглобина). [6] Дисбаланс альфа- и бета-цепей глобина приводит к неэффективному эритропоэзу , повышенному гемолизу и нарушенному гомеостазу железа . [7] Пациентам могут потребоваться повторные переливания крови на протяжении всей жизни для поддержания достаточного уровня гемоглобина. Следовательно, у пациентов также могут возникнуть серьезные проблемы, связанные с перегрузкой железом . [4]
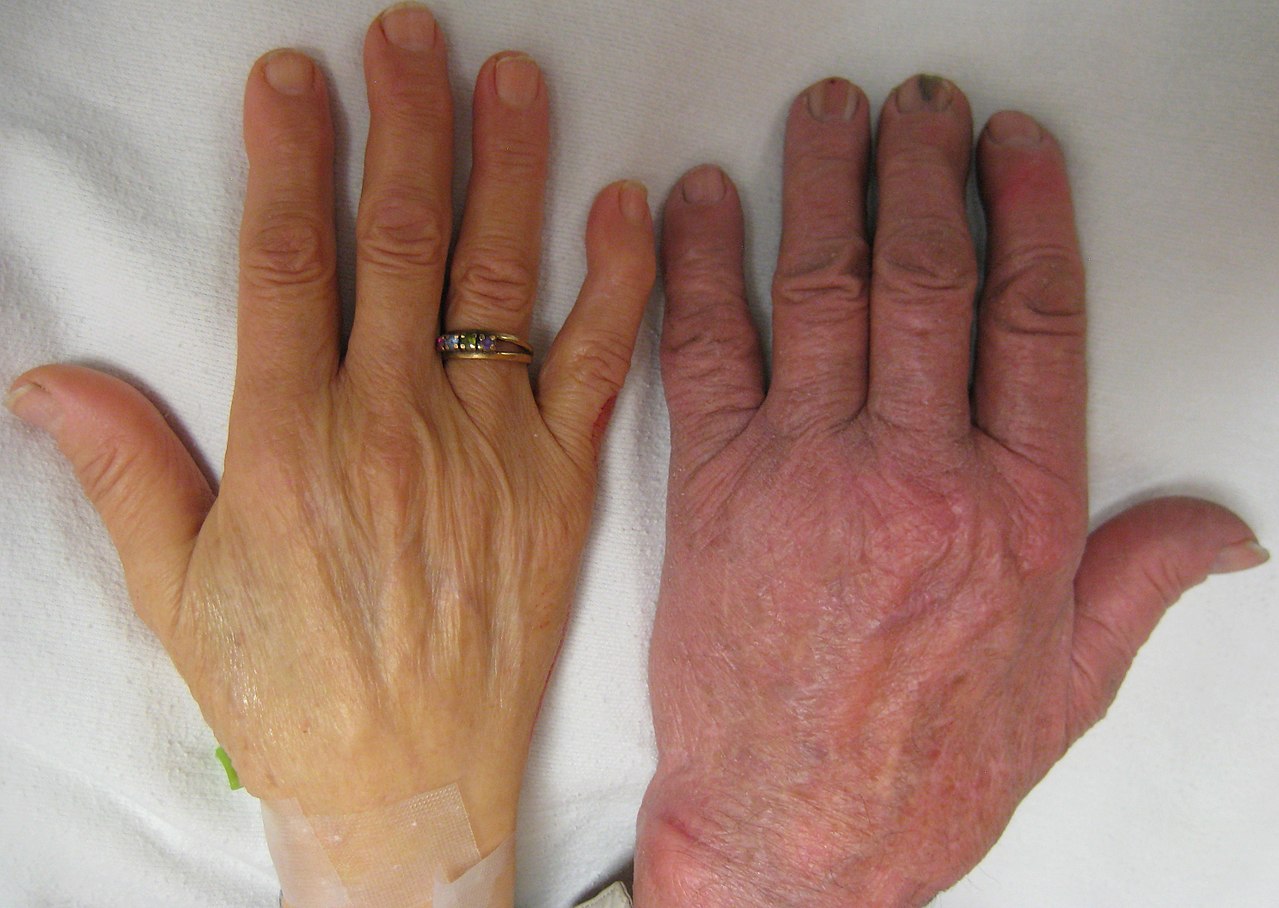
Описаны три основные формы: малая талассемия, промежуточная талассемия и большая талассемия, которые варьируются от бессимптомных или легких симптомов до тяжелой анемии, требующей пожизненных переливаний. [8] У людей с большой бета-талассемией (те, кто гомозиготен по мутациям талассемии или унаследовал 2 мутации) обычно в течение первых двух лет жизни проявляется симптоматическая тяжелая анемия, плохой рост и скелетные аномалии. Нелеченная большая талассемия в конечном итоге приводит к смерти, обычно из-за сердечной недостаточности ; поэтому пренатальный скрининг очень важен. [9] У людей с промежуточной бета-талассемией (те, кто является сложными гетерозиготами по мутации бета-талассемии) обычно в более позднем возрасте проявляются легкие или умеренные симптомы анемии. [8] Признак бета-талассемии (также известный как малая бета-талассемия) включает гетерозиготное наследование мутации бета-талассемии, и у пациентов обычно наблюдается микроцитоз с пограничной гипохромной анемией, и они обычно бессимптомны или имеют легкие симптомы. [8] Малая бета-талассемия может также проявляться как молчаливые носители бета-талассемии; те, кто наследует мутацию бета-талассемии, но не имеет гематологических отклонений или симптомов. [8] Некоторые люди с талассемией подвержены осложнениям со здоровьем, которые затрагивают селезенку (гиперспленизм) и желчные камни (из-за гипербилирубинемии от периферического гемолиза). [8] [1] Эти осложнения в основном встречаются у пациентов с большой и промежуточной талассемией. [ необходима ссылка ]
Избыток железа (от гемолиза или переливания ) вызывает серьезные осложнения в печени, сердце и эндокринных железах . Тяжелые симптомы включают цирроз печени , фиброз печени и, в крайних случаях, рак печени . [10] Сердечная недостаточность, нарушение роста, диабет и остеопороз являются опасными для жизни состояниями, которые могут быть вызваны большой бета-талассемией. [11] Основные сердечные аномалии, наблюдаемые в результате бета-талассемии и перегрузки железом, включают систолическую и диастолическую дисфункцию левого желудочка, легочную гипертензию, вальвулопатию, аритмии и перикардит. Повышенное всасывание железа в желудочно-кишечном тракте наблюдается при всех стадиях бета-талассемии, а повышенное разрушение эритроцитов селезенкой из-за неэффективного эритропоэза дополнительно высвобождает дополнительное железо в кровоток . [12]
Дополнительные симптомы большой или средней бета-талассемии включают классические симптомы умеренной или тяжелой анемии, включая усталость, задержку роста и развития в детстве, язвы ног и недостаточность органов. [8] Неэффективный эритропоэз (выработка эритроцитов) также может привести к компенсаторному расширению костного мозга, что затем может привести к костным изменениям/деформациям, болям в костях и краниофациальным аномалиям. [8] Экстрамедуллярные органы, такие как печень и селезенка, которые также могут подвергаться эритропоэзу, активируются, что приводит к гепатоспленомегалии (увеличению печени и селезенки). [8] Другие ткани в организме также могут стать местами эритропоэза, что приводит к экстрамедуллярным гемопоэтическим псевдоопухолям, которые могут вызывать компрессионные симптомы, если они возникают в грудной полости или позвоночном канале. [8]
Можно выделить две основные группы мутаций:
Мутации характеризуются как (βo), если они предотвращают любое образование β-глобиновых цепей, мутации характеризуются как (β+), если они допускают некоторое образование β-глобиновых цепей. [8]
Для принятия решений по управлению и соответствия требованиям клинических испытаний используются категории трансфузионно-зависимой талассемии (TDT) и нетрансфузионно-зависимой талассемии (NTDT). Пациенты обычно считаются имеющими NTDT, если они получили менее 6 единиц эритроцитов за последние 6 месяцев и ни одной за предыдущие 2 месяца. [18]

Бета-талассемия — наследственное заболевание, поражающее гемоглобин. Как и в случае примерно половины всех наследственных заболеваний, [19] наследственная мутация повреждает сборку РНК-мессенджера (мРНК), которая транскрибируется с хромосомы . ДНК содержит как инструкции ( гены ) для связывания аминокислот в белки , так и участки ДНК, которые играют важную роль в регуляции уровней продуцируемых белков . [20]
При талассемии в мРНК включается дополнительная, непрерывная длина или прерывистый фрагмент некодирующих инструкций. Это происходит, потому что мутация стирает границу между интронными и экзонными частями ДНК-матрицы. [21] Поскольку все кодирующие секции могут все еще присутствовать, может быть произведен нормальный гемоглобин, а добавленный генетический материал, если он вызывает патологию, вместо этого нарушает регуляторные функции в достаточной степени, чтобы вызвать анемию. Нормальные альфа- и бета-субъединицы гемоглобина имеют железосодержащую центральную часть (гем), которая позволяет белковой цепи субъединицы сворачиваться вокруг нее. Нормальный взрослый гемоглобин содержит 2 альфа- и 2 бета-субъединицы. [22] Талассемии обычно затрагивают только мРНК для производства бета-цепей (отсюда и название). Поскольку мутация может быть изменением только одного основания ( однонуклеотидный полиморфизм ), текущие усилия направлены на поиск генной терапии для внесения этой единственной коррекции. [23] [24]
Семейный анамнез и происхождение являются факторами, которые увеличивают риск бета-талассемии. В зависимости от семейного анамнеза, если у родителей или бабушек и дедушек человека была большая или промежуточная бета-талассемия, существует 75% (3 из 4) вероятности (см. таблицу наследования вверху страницы) того, что мутировавший ген будет унаследован потомством. Даже если у ребенка нет большой или промежуточной бета-талассемии, он все равно может быть носителем, что может привести к тому, что будущие поколения его потомков будут иметь бета-талассемию. [ необходима цитата ]
Другим фактором риска является происхождение. Бета-талассемия чаще всего встречается у людей итальянского, греческого, ближневосточного, южноазиатского и африканского происхождения. [25]

Боль в животе из-за гиперспленизма , инфаркт селезенки и боль в правом верхнем квадранте, вызванная желчными камнями, являются основными клиническими проявлениями. Однако диагностика талассемии только по симптомам недостаточна. Врачи отмечают эти признаки как ассоциативные из-за сложности этого заболевания. [26] Следующие ассоциативные признаки могут свидетельствовать о тяжести фенотипа : бледность, плохой рост, недостаточное потребление пищи, спленомегалия , желтуха , гиперплазия верхней челюсти, неправильный прикус зубов , желчнокаменная болезнь , систолический шум выброса при наличии тяжелой анемии и патологических переломов. На основании симптомов назначаются тесты для дифференциальной диагностики . Эти тесты включают общий анализ крови ; электрофорез гемоглобина ; сывороточный трансферрин , ферритин , общую железосвязывающую способность ; уробилин и уробилоген мочи ; мазок периферической крови , который может показать кодоциты или клетки-мишени; [27] гематокрит ; и сывороточный билирубин. [28] [29] Ожидаемая картина при электрофорезе гемоглобина у людей с бета-талассемией - повышенный уровень гемоглобина А2 и слегка повышенный уровень гемоглобина F. [ необходима цитата ] Диагноз подтверждается с помощью электрофореза гемоглобина или высокоэффективной жидкостной хроматографии. [8]
Изменения скелета, связанные с расширением костного мозга:
Все бета-талассемии могут иметь аномальные эритроциты; семейный анамнез отслеживается с помощью анализа ДНК. [3] Этот тест используется для исследования делеций и мутаций в генах, продуцирующих альфа- и бета-глобин. Семейные исследования могут быть проведены для оценки статуса носителя и типов мутаций, присутствующих у других членов семьи. Тестирование ДНК не является рутинным, но может помочь диагностировать талассемию и определить статус носителя. В большинстве случаев лечащий врач использует клиническую предварительную диагностику, оценивая симптомы анемии: усталость, одышку и плохую переносимость физических нагрузок. [30] Дальнейший генетический анализ может включать ВЭЖХ , если обычный электрофорез окажется затруднительным. [28]
Бета-талассемия — наследственное заболевание, допускающее профилактическое лечение путем скрининга носителей и пренатальной диагностики. Его можно предотвратить, если у одного из родителей нормальные гены, что дает начало скринингам, которые позволяют носителям выбирать партнеров с нормальным гемоглобином. Исследование, направленное на выявление генов, которые могут дать начало потомству с серповидноклеточной анемией. Пациенты с диагнозом бета-талассемия имеют MCH ≤ 26 пг и RDW < 19. Из 10 148 пациентов 1739 пациентов имели фенотип гемоглобина и RDW, соответствующие бета-талассемии. После сужения пациентов были протестированы уровни HbA2, у 77 пациентов была бета-талассемия. [31] Эта процедура скрининга оказалась нечувствительной в популяциях западноафриканского происхождения из-за показателей высокой распространенности альфа-талассемии. В странах существуют программы распространения информации о репродуктивных рисках, связанных с носителями гемоглобинопатий. Программы скрининга носителей талассемии включают образовательные программы в школах, вооруженных силах и через средства массовой информации, а также консультирование носителей и пар носителей. [32] Скрининг показал снижение заболеваемости; к 1995 году распространенность в Италии снизилась с 1:250 до 1:4000, и на 95% снизилась в этом регионе. Снижение заболеваемости пошло на пользу тем, кто страдает талассемией, поскольку спрос на кровь снизился, следовательно, улучшилось снабжение лечением. [ необходима цитата ]

Больным детям требуются регулярные пожизненные переливания крови . Трансплантация костного мозга может быть излечивающей для некоторых детей. [33] Пациенты получают частые переливания крови, которые приводят к перегрузке железом или усиливают ее . [34] Лечение хелатированием железа необходимо для предотвращения повреждения внутренних органов в случаях перегрузки железом. Достижения в лечении хелатированием железа позволяют пациентам с большой талассемией жить долго, имея доступ к надлежащему лечению. Популярные хелаторы включают дефероксамин и деферипрон . [35] [36]
Пероральный хелатор деферазирокс был одобрен для использования в 2005 году в некоторых странах. [37] [38] Трансплантация костного мозга является единственным лекарством и показана пациентам с тяжелой большой талассемией. Трансплантация может устранить зависимость пациента от переливаний. При отсутствии соответствующего донора, брат или сестра-спаситель могут быть зачаты с помощью предимплантационной генетической диагностики (ПГД), чтобы быть свободными от заболевания, а также соответствовать типу человеческого лейкоцитарного антигена (HLA) реципиента . [39]
Сывороточный ферритин (форма хранения железа) обычно измеряется у пациентов с бета-талассемией для определения степени перегрузки железом; при повышенном уровне ферритина назначается терапия хелаторами железа. Три хелатора железа; подкожный дефероксамин, пероральный деферипрон и пероральный деферазирокс могут использоваться в качестве монотерапии или в комбинации; все они, как было показано, снижают сывороточный/системный уровень железа, печеночный и сердечный уровень железа, а также снижают риск сердечной аритмии, сердечной недостаточности и смерти. [8] МРТ печени и миокарда также используется для количественной оценки отложения железа в целевых органах, особенно в сердце и печени, для определения терапии. [8]
Ученые из Медицинского колледжа Вейлла Корнелла разработали стратегию генной терапии, которая может реально лечить как бета-талассемию, так и серповидноклеточную анемию. Технология основана на доставке лентивирусного вектора, несущего как ген человеческого β-глобина, так и инсулятор анкирина для улучшения транскрипции и трансляции генов, а также повышения уровня продукции β-глобина. [40]
10 июня 2022 года федеральная консультативная группа США рекомендовала FDA одобрить генную терапию для использования при бета-талассемии. [41] Производитель Bluebird bio взимает в Соединенных Штатах 2,8 миллиона долларов за одноразовое лечение Zynteglo ( betibeglogene autotemcel ). [42] [43]
Разработаны методы генной терапии, направленные на увеличение продукции фетального гемоглобина при бета-талассемии, а также при серповидноклеточной анемии путем ингибирования гена BCL11A . [44] [45] Exagamglogene autotemcel , продаваемый под торговой маркой Casgevy, представляет собой генную терапию для лечения бета-талассемии, зависящей от переливания крови, разработанную Vertex Pharmaceuticals и CRISPR Therapeutics . [46]
Лечение было одобрено в Соединенном Королевстве для лечения бета-талассемии, зависящей от переливания крови, в ноябре 2023 года [47] [48] [49] и в Соединенных Штатах в январе 2024 года. [50] [51] [52]
Exagamglogene autotemcel — это первый метод генной терапии на основе клеток, использующий технологию редактирования генов CRISPR/Cas9, одобренный Управлением по контролю за продуктами и лекарствами США (FDA). [50]
Генная терапия производится из собственных стволовых клеток крови реципиента, которые модифицируются и возвращаются в виде одноразовой инфузии одной дозы в рамках трансплантации гемопоэтических стволовых клеток . Перед лечением собираются собственные стволовые клетки реципиента, а затем реципиент должен пройти миелоаблативное кондиционирование (высокодозную химиотерапию), процесс, который удаляет клетки из костного мозга, чтобы их можно было заменить модифицированными клетками в exagamglogene autotemcel . [ необходима цитата ]
Пациенты с большой талассемией более склонны к спленэктомии. Использование спленэктомии в последние годы снижается из-за снижения распространенности гиперспленизма у пациентов, которым адекватно переливают кровь. Спленэктомия также связана с повышенным риском инфекций и повышенной заболеваемостью из-за сосудистых заболеваний, поскольку селезенка участвует в очистке организма от патологических или аномальных эритроцитов. [8] Пациенты с гиперспленизмом с большей вероятностью имеют меньшее количество здоровых клеток крови в организме, чем обычно, и проявляют симптомы анемии. Различными хирургическими методами являются открытый и лапароскопический методы. [2] Лапароскопический метод требует более длительного времени операции, но более короткого периода восстановления с меньшим и менее заметным хирургическим рубцом. Если нет необходимости удалять всю селезенку, может быть проведена частичная спленэктомия; этот метод сохраняет часть иммунной функции, одновременно снижая вероятность гиперспленизма. Лица, которым предстоит спленэктомия, должны получить соответствующую пневмококковую вакцину как минимум за одну неделю (предпочтительно за три недели) до операции. [53]
Долгосрочная трансфузионная терапия (у пациентов с трансфузионно-зависимой бета-талассемией) — это лечение, используемое для поддержания уровня гемоглобина на целевом предтрансфузионном уровне 9–10,5 г/дл (11–12 г/дл у пациентов с сопутствующими заболеваниями сердца). [8] Для обеспечения качественного переливания крови эритроцитарная масса должна быть лейкоредуцирована. При наличии лейкоредуцированных пакетов крови у пациента снижается риск развития побочных реакций из-за загрязненных лейкоцитов и предотвращается аллоиммунизация тромбоцитов. [54] Пациенты с аллергическими реакциями на переливание или необычными антителами к эритроцитам должны получать промытые эритроциты или криоконсервированные эритроциты. Промытые эритроциты были удалены из плазменных белков, которые могли бы стать мишенью для антител пациента, что позволяет безопасно проводить переливание. Криоконсервированные эритроциты используются для поддержания запаса редких донорских единиц для пациентов с необычными антителами к эритроцитам или отсутствующими общими антигенами эритроцитов. Эти регулярные переливания способствуют нормальному росту и физической активности, а также подавляют гиперактивность костного мозга и экстрамедуллярное кроветворение, помогая в некоторых случаях уменьшить болезненные массы. [18] Однако эти преимущества следует сопоставлять с соображениями перегрузки железом, и по мере прогрессирования клинического течения следует вносить коррективы. [18]
При нормальном гомеостазе железа циркулирующее железо связано с трансферрином. Но при перегрузке железом (например, при частых переливаниях крови) способность трансферрина связывать железо превышается, и накапливается несвязанное с трансферрином железо. Это несвязанное железо токсично из-за его высокой склонности вызывать формы кислорода и отвечает за повреждение клеток. Профилактика перегрузки железом защищает пациентов от заболеваемости и смертности. Основная цель — связать и вывести железо из организма со скоростью, равной скорости поступления трансфузионного железа или превышающей поступление железа. [55] Хелатирование железа — это медицинская терапия, которая может предотвратить осложнения перегрузки железом. [8] Каждая единица перелитой крови содержит 200–250 мг железа, и у организма нет естественного механизма для удаления избытка железа. Избыток железа можно удалить с помощью хелаторов железа (дефероксамин, деферипрон и деферазирокс). [56] Концентрация железа в печени (LIC) может быть измерена с помощью магнитно-резонансной томографии (МРТ) R2 или T2* . В тех частях мира, где нет современных методов МРТ, в качестве альтернативы можно использовать сывороточный ферритин . У пациентов с перегрузкой железом разумно начинать хелатирующую терапию, если LIC >5 мг Fe на грамм сухого веса (dw) или уровень сывороточного ферритина >800 нг/мл. Хелатирующую терапию можно прекратить, если LIC <3 мг Fe на грамм сухого веса или сывороточный ферритин <300 нг/мл. [18]
Luspatercept (ACE-536) — это рекомбинантный слитый белок, который используется в качестве лечения взрослых с трансфузионно-зависимой бета-талассемией. Он состоит из модифицированного внеклеточного домена человеческого рецептора активина типа IIB, связанного с Fc-частью человеческого антитела IgG1. [8] Молекула связывается с выбранными лигандами суперсемейства трансформирующего фактора роста бета, чтобы блокировать сигнализацию SMAD2 и 3 , тем самым усиливая созревание эритроидов. [8] Было показано, что препарат снижает нагрузку на переливание на 33% у взрослых с трансфузионно-зависимой бета-талассемией по сравнению с плацебо, а также был связан со снижением уровня ферритина (без существенного снижения уровня железа в печени или сердце). [8]
Пациенты с промежуточной бета-талассемией обычно проявляются в возрасте >2 лет с легкой или умеренной анемией (7-10 г/дл). [18] Им может не потребоваться переливание крови или могут потребоваться эпизодические переливания крови при определенных обстоятельствах (инфекция, беременность, операция). [8] У пациентов с частыми переливаниями может развиться перегрузка железом , и им потребуется хелатная терапия . [57] Передача аутосомно-рецессивная ; однако сообщалось о доминантных мутациях и сложных гетерозиготах . Рекомендуется генетическое консультирование и может быть предложена пренатальная диагностика . [58]
Пациенты с малой бета-талассемией обычно не имеют симптомов и часто наблюдаются без лечения. [8] Малая бета-талассемия может сосуществовать с другими состояниями, такими как хронический гепатит B , хронический гепатит C , неалкогольная жировая болезнь печени и алкогольная болезнь печени , которые при сочетании или сосуществовании могут привести к перегрузке печени железом и более тяжелому заболеванию печени. [59]
Бета-форма талассемии особенно распространена среди средиземноморских народов, и эта географическая ассоциация ответственна за ее название: thalassa ( θάλασσα ) — греческое слово, обозначающее море, а haima ( αἷμα ) — греческое слово, обозначающее кровь. [60] [61] [ требуется цитирование ] В Европе самые высокие концентрации заболевания обнаружены в Греции и прибрежных районах Турции . Основные средиземноморские острова (за исключением Балеарских ), такие как Сицилия , Сардиния , Корсика , Кипр , Мальта и Крит , особенно сильно поражены. [62] [63] Другие средиземноморские народы , а также те, которые находятся в непосредственной близости от Средиземного моря, также имеют высокие показатели заболеваемости, включая людей из Западной Азии и Северной Африки . В Пакистане 6% от общей численности населения являются носителями бета-талассемии из-за самого высокого уровня кузенских браков в мире. Данные показывают, что 15% греков и турок -киприотов являются носителями генов бета-талассемии, в то время как 10% населения являются носителями генов альфа-талассемии . [64]
Талассемия может давать степень защиты от малярии , [65] которая распространена или была распространена в регионах, где распространена эта черта, тем самым предоставляя избирательное преимущество выживания носителям (известное как гетерозиготное преимущество ), таким образом, увековечивая мутацию. В этом отношении различные талассемии напоминают другое генетическое заболевание, влияющее на гемоглобин, серповидноклеточную анемию . [66]
Расстройство более распространено среди определенных этнических групп и возрастных групп. Бета-талассемия наиболее распространена в «поясе талассемии», который включает районы Африки к югу от Сахары, Средиземноморья, простирающегося до Ближнего Востока и Юго-Восточной Азии. [8] Считается, что такое географическое распределение обусловлено носительством бета-талассемии (малая бета-талассемия), дающим устойчивость к малярии. [8] В Соединенных Штатах распространенность талассемии составляет приблизительно 1 на 272 000 или 1 000 человек. В 2002 году в Англии было зарегистрировано 4 000 госпитализированных случаев и 9 233 эпизода консультаций по поводу талассемии. Мужчины составили 53% эпизодов консультаций в больнице, а женщины — 47%. Средний возраст пациентов составляет 23 года, и только 1% консультантов были старше 75 лет, а 69% — в возрасте 15–59 лет. По оценкам, 1,5% населения мира являются носителями, и ежегодно рождается 40 000 больных младенцев. [8] Большая бета-талассемия обычно приводит к летальному исходу в младенчестве, если немедленно не начать переливание крови. [67]